
An Overview of the TRP-Oxidative Stress Axis in Metabolic Syndrome: Insights for Novel Therapeutic Approaches

 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Adiponectin Dysregulation, Oxidative Stress and Inflammation as Mechanisms of Metabolic Syndrome
2.1. Adiponectin Dysregulation
2.2. Oxidative Stress
2.3. Inflammation
3. Transient Receptor Potential Channels
3.1. General Overview of TRPV1, TRPA1 and TRPC5 Channels
3.2. TRPs as Key Sensors of Oxidative Stress
3.3. TRPs as Regulators of Inflammation
4. The Roles of TRPV1, TRPA1 and TRPC5 in MS
4.1. Regulation of Insulin and Insulin Resistance
4.2. Regulation of Adypocytes
4.3. TRPs and the Liver
4.4. TRPs and Skeletal Muscle
4.5. Connecting Metabolic Tissues and the Central Nervous System
5. Clinical Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory Mechanisms Linking Obesity and Metabolic Disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic Syndrome: Therapeutic Considerations; Springer: Berlin/Heidelberg, Germany, 2005; Volume 170. [Google Scholar] [CrossRef]
- Altabas, V. Drug Treatment of Metabolic Syndrome. Curr. Clin. Pharmacol. 2016, 8, 224–231. [Google Scholar] [CrossRef]
- Barroso, I.; McCarthy, M.I. The Genetic Basis of Metabolic Disease. Cell 2019, 177, 146–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Ahn, J.; Park, J.; Na, H.; Lee, Y.; Kim, Y.; Hong, G.; Lee, K.-R. Genetic Diversity of Insulin Resistance and Metabolic Syndrome; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Misselbeck, K.; Parolo, S.; Lorenzini, F.; Savoca, V.; Leonardelli, L.; Bora, P.; Morine, M.J.; Mione, M.C.; Domenici, E.; Priami, C. A Network-Based Approach to Identify Deregulated Pathways and Drug Effects in Metabolic Syndrome. Nat. Commun. 2019, 10, 5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Frankenberg, A.D.; Reis, A.F.; Gerchman, F. Relationships between Adiponectin Levels, the Metabolic Syndrome, and Type 2 Diabetes: A Literature Review. Arch. Endocrinol. Metab. 2017, 61, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic Syndrome: Pathophysiology, Management, and Modulation by Natural Compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargar, B.; Zamanian, Z.; Hosseinabadi, M.B.; Gharibi, V.; Moradi, M.S.; Cousins, R. Understanding the Role of Oxidative Stress in the Incidence of Metabolic Syndrome and Obstructive Sleep Apnea. BMC Endocr. Disord. 2021, 21, 77. [Google Scholar] [CrossRef]
- Colak, E.; Pap, D. The Role of Oxidative Stress in the Development of Obesity and Obesity-Related Metabolic Disorders. J. Med. Biochem. 2021, 40, 1–9. [Google Scholar] [CrossRef]
- Numata, T.; Takahashi, K.; Inoue, R. “TRP Inflammation” Relationship in Cardiovascular System. Semin. Immunopathol. 2016, 38, 339–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Takeda, T.-A.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological Roles of Zinc Transporters: Molecular and Genetic Importance in Zinc Homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Luo, Y.; Zhang, F.; Tang, S.; Zhu, T. Involvement of TRP Channels in Adipocyte Thermogenesis: An Update. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Hellenthal, K.E.M.; Brabenec, L.; Gross, E.R.; Wagner, N.M. TRP Channels as Sensors of Aldehyde and Oxidative Stress. Biomolecules 2021, 11, 1401. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A Novel Serum Protein Similar to C1q, Produced Exclusively in Adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Okubo, K.; Shimomura, I.; Funahashi, T.; Matsuzawa, Y.; Matsubara, K. CDNA Cloning and Expression of a Novel Adipose Specific Collagen-like Factor, ApM1 (Adipose Most Abundant Gene Transcript 1). Biochem. Biophys. Res. Commun. 1996, 221, 286–289. [Google Scholar] [CrossRef]
- Nakano, Y.; Tobe, T.; Choi-Miura, N.-H.; Mazda, T.; Tomita, M. Isolation and Characterization of GBP28, a Novel Gelatin-Binding Protein Purified from Human Plasma. J. Biochem. 1996, 120, 803–812. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Hawkins, M.; Combs, T.P.; Rajala, M.W.; Doebber, T.; Berger, J.P.; Wagner, J.A.; Wu, M.; Knopps, A.; Xiang, A.H.; et al. Complex Distribution, Not Absolute Amount of Adiponectin, Correlates with Thiazolidinedione-Mediated Improvement in Insulin Sensitivity. J. Biol. Chem. 2004, 279, 12152–12162. [Google Scholar] [CrossRef] [Green Version]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Schere, P.E. The Adipocyte-Secreted Protein Acrp30 Enhances Hepatic Insulin Action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Combs, T.P.; Berg, A.H.; Obici, S.; Scherer, P.E.; Rossetti, L. Endogenous Glucose Production Is Inhibited by the Adipose-Derived Protein Acrp30. J. Clin. Investig. 2001, 108, 1875–1881. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The Fat-Derived Hormone Adiponectin Reverses Insulin Resistance Associated with Both Lipoatrophy and Obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Takahashi, N.; Hileman, S.M.; Patel, H.R.; Berg, A.H.; Pajvani, U.B.; Scherer, P.E.; Ahima, R.S. Adiponectin Acts in the Brain to Decrease Body Weight. Nat. Med. 2004, 10, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.B.; Somwar, R.; Maida, A.; Fang, X.; Bikopoulos, G.; Sweeney, G. Globular Adiponectin Increases GLUT4 Translocation and Glucose Uptake but Reduces Glycogen Synthesis in Rat Skeletal Muscle Cells. Diabetologia 2005, 48, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Palanivel, R.; Zhou, X.; Liu, Y.; Xu, A.; Wang, Y.; Sweeney, G. Hyperglycemia- and Hyperinsulinemia-Induced Alteration of Adiponectin Receptor Expression and Adiponectin Effects in L6 Myoblasts. J. Mol. Endocrinol. 2005, 35, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruebis, J.; Tsao, T.-S.; Javorschi, S.; Ebbets-Reed, D.; Ruth, M.; Erickson, S.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic Cleavage Product of 30-KDa Adipocyte Complement-Related Protein Increases Fatty Acid Oxidation in Muscle and Causes Weight Loss in Mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, H.; Oritani, K.; Kato, H.; Yokota, T.; Takahashi, I.; Maeda, T.; Masaie, H.; Ichii, M.; Kamada, Y.; Tamura, S.; et al. Identification of Amino-Terminal Region of Adiponectin as a Physiologically Functional Domain. J. Cell. Biochem. 2006, 98, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Okada-Iwabu, M.; Yamauchi, T.; Kadowaki, T. Adiponectin/AdipoR Research and Its Implications for Lifestyle-Related Diseases. Front. Cardiovasc. Med. 2019, 6, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thundyil, J.; Pavlovski, D.; Sobey, C.G.; Arumugam, T.V. Adiponectin Receptor Signalling in the Brain. Br. J. Pharmacol. 2012, 165, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Bjursell, M.; Ahnmark, A.; Bohlooly-Y, M.; William-Olsson, L.; Rhedin, M.; Peng, X.R.; Ploj, K.; Gerdin, A.K.; Arnerup, G.; Elmgren, A.; et al. Opposing Effects of Adiponectin Receptors 1 and 2 on Energy Metabolism. Diabetes 2007, 56, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Cinti, S.; Frederich, R.C.; Zingaretti, M.C.; de Matteis, R.; Flier, J.S.; Lowell, B.B. Immunohistochemical Localization of Leptin and Uncoupling Protein in White and Brown Adipose Tissue. Endocrinology 1997, 138, 797–804. [Google Scholar] [CrossRef]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-Induced Insulin Resistance in Mice Lacking Adiponectin/ACRP30. Nat. Med. 2002, 8, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Salmenniemi, U.; Ruotsalainen, E.; Pihlajamäki, J.; Vauhkonen, I.; Kainulainen, S.; Punnonen, K.; Vanninen, E.; Laakso, M. Multiple Abnormalities in Glucose and Energy Metabolism and Coordinated Changes in Levels of Adiponectin, Cytokines, and Adhesion Molecules in Subjects with Metabolic Syndrome. Circulation 2004, 110, 3842–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumada, M.; Kihara, S.; Sumitsuji, S.; Kawamoto, T.; Matsumoto, S.; Ouchi, N.; Arita, Y.; Okamoto, Y.; Shimomura, I.; Hiraoka, H.; et al. Association of Hypoadiponectinemia with Coronary Artery Disease in Men. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, M.; Shimomura, I.; Sata, M.; Arita, Y.; Nishida, M.; Maeda, N.; Kumada, M.; Okamoto, Y.; Nagaretani, H.; Nishizawa, H.; et al. Role of Adiponectin in Preventing Vascular Stenosis. The Missing Link of Adipo-Vascular Axis. J. Biol. Chem. 2002, 277, 37487–37491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollias, A.; Tsiotra, P.C.; Ikonomidis, I.; Maratou, E.; Mitrou, P.; Kyriazi, E.; Boutati, E.; Lekakis, J.; Economopoulos, T.; Kremastinos, D.T.; et al. Adiponectin Levels and Expression of Adiponectin Receptors in Isolated Monocytes from Overweight Patients with Coronary Artery Disease. Cardiovasc. Diabetol. 2011, 10, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, A.F.; Guichardant, M.; Cozzone, D.; Bernoud-Hubac, N.; Bouzaïdi-Tiali, N.; Lagarde, M.; Géloën, A. Effects of Oxidative Stress on Adiponectin Secretion and Lactate Production in 3T3-L1 Adipocytes. Free Radic. Biol. Med. 2005, 38, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lam, K.S.L.; Wang, Y.; Wu, D.; Lam, M.C.; Shen, J.; Wong, L.; Hoo, R.L.C.; Zhang, J.; Xu, A. Hypoxia Dysregulates the Production of Adiponectin and Plasminogen Activator Inhibitor-1 Independent of Reactive Oxygen Species in Adipocytes. Biochem. Biophys. Res. Commun. 2006, 341, 549–556. [Google Scholar] [CrossRef]
- Monickaraj, F.; Aravind, S.; Nandhini, P.; Prabu, P.; Sathishkumar, C.; Mohan, V.; Balasubramanyam, M. Accelerated Fat Cell Aging Links Oxidative Stress and Insulin Resistance in Adipocytes. J. Biosci. 2013, 38, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Kowalska, K.; Olejnik, A. Cranberries (Oxycoccus quadripetalus) Inhibit pro-Inflammatory Cytokine and Chemokine Expression in 3T3-L1 Adipocytes. Food Chem. 2016, 196, 1137–1143. [Google Scholar] [CrossRef]
- Fukushima, M.; Okamoto, Y.; Katsumata, H.; Ishikawa, M.; Ishii, S.; Okamoto, M.; Minami, S. Growth Hormone Ameliorates Adipose Dysfunction during Oxidative Stress and Inflammation and Improves Glucose Tolerance in Obese Mice. Horm. Metab. Res. 2014, 46, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Qiao, Q.Y.; Pan, L.H.; Zhou, D.C.; Hu, C.; Gu, H.F.; Fu, S.K.; Liu, X.L.; Jin, H.M. Losartan Reduces Insulin Resistance by Inhibiting Oxidative Stress and Enhancing Insulin Signaling Transduction. Exp. Clin. Endocrinol. Diabetes 2015, 123, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Schroeder, P.; Sies, H. Peroxynitrite Signaling: Receptor Tyrosine Kinases and Activation of Stress-Responsive Pathways. Free Radic. Biol. Med. 2002, 33, 737–743. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxidative Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 8267234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, J.; Galano, J.M.; Durand, T.; le Guennec, J.Y.; Lee, J.C.Y. Physiological Role of Reactive Oxygen Species as Promoters of Natural Defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef] [Green Version]
- Oguntibeju, O.O. Type 2 Diabetes Mellitus, Oxidative Stress and Inflammation: Examining the Links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45. [Google Scholar]
- Cheng, X.; Siow, R.C.M.; Mann, G.E. Impaired Redox Signaling and Antioxidant Gene Expression in Endothelial Cells in Diabetes: A Role for Mitochondria and the Nuclear Factor-E2-Related Factor 2-Kelch-Like ECH-Associated Protein 1 Defense Pathway. Antioxid. Redox Signal. 2011, 14, 469–487. [Google Scholar] [CrossRef]
- Foster, M.W.; McMahon, T.J.; Stamler, J.S. S-Nitrosylation in Health and Disease. Trends Mol. Med. 2003, 9, 160–168. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos-Morón, E.; Abad-Jiménez, Z.; de Marañón, A.M.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezza, T.; de Marañón, A.M.; Canet, F.; Díaz-Pozo, P.; Marti, M.; D’ocon, P.; Apostolova, N.; Rocha, M.; Víctor, V.M. MicroRNAs and Oxidative Stress: An Intriguing Crosstalk to Be Exploited in the Management of Type 2 Diabetes. Antioxidants 2021, 10, 802. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, J.L.; Zoukourian, C.; Chappey, O.; Wautier, M.P.; Guillausseau, P.J.; Cao, R.; Horl, O.; Stern, D.; Schmidt, A.M. Receptor-Mediated Endothelial Cell Dysfunction in Diabetic Vasculopathy. Soluble Receptor for Advanced Glycation End Products Blocks Hyperpermeability in Diabetic Rats. J. Clin. Investig. 1996, 97, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Berwick, Z.C.; Dick, G.M.; Tune, J.D. Heart of the Matter: Coronary Dysfunction in Metabolic Syndrome. J. Mol. Cell. Cardiol. 2012, 52, 848–856. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, R.; Yan, S.F.; Herold, K.; Clynes, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products: Fundamental Roles in the Inflammatory Response: Winding the Way to the Pathogenesis of Endothelial Dysfunction and Atherosclerosis. Ann. N. Y. Acad. Sci. 2008, 1126, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Huth, C.; Pigeon, É.; Riou, M.È.; St-Onge, J.; Arguin, H.; Couillard, E.; Dubois, M.J.; Marette, A.; Tremblay, A.; Weisnagel, S.J.; et al. Fitness, Adiposopathy, and Adiposity Are Independent Predictors of Insulin Sensitivity in Middle-Aged Men without Diabetes. J. Physiol. Biochem. 2016, 72, 435–444. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Ronald Kahn, C. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose Tissue Heterogeneity: Implication of Depot Differences in Adipose Tissue for Obesity Complications. Mol. Asp. Med. 2013, 34, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Kakisaka, Y.; Nakashima, T.; Sumida, Y.; Yoh, T.; Nakamura, H.; Yodoi, J.; Senmaru, H. Elevation of Serum Thioredoxin Levels in Patients with Type 2 Diabetes. Horm. Metab. Res. 2002, 34, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Kishimoto, C.; Nakamura, H.; Makita, T.; Ishii, K.; Okuda, N.; Yodoi, J.; Sasayama, S. Serum Thioredoxin and α-Tocopherol Concentrations in Patients with Major Risk Factors. Circ. J. 2005, 69, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallmeier, D.; Larson, M.G.; Vasan, R.S.; Keaney, J.F.; Fontes, J.D.; Meigs, J.B.; Fox, C.S.; Benjamin, E.J. Metabolic Syndrome and Inflammatory Biomarkers: A Community-Based Cross-Sectional Study at the Framingham Heart Study. Diabetol. Metab. Syndr. 2012, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Kim, C.S.; Kang, J.H. Inflammatory Components of Adipose Tissue as Target for Treatment of Metabolic Syndrome. Forum Nutr. 2009, 61, 95–103. [Google Scholar] [CrossRef]
- Rastelli, M.; Knauf, C.; Cani, P.D. Gut Microbes and Health: A Focus on the Mechanisms Linking Microbes, Obesity, and Related Disorders. Obesity 2018, 26, 792–800. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Patel, V.H. Inflammation and Metabolic Syndrome: An Overview. Curr. Res. Nutr. Food Sci. 2015, 3, 263–268. [Google Scholar] [CrossRef]
- Collins, K.H.; Herzog, W.; MacDonald, G.Z.; Reimer, R.A.; Rios, J.L.; Smith, I.C.; Zernicke, R.F.; Hart, D.A. Obesity, Metabolic Syndrome, and Musculoskeletal Disease: Common Inflammatory Pathways Suggest a Central Role for Loss of Muscle Integrity. Front. Physiol. 2018, 9, 112. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The Effects of Palmitate on Hepatic Insulin Resistance Are Mediated by NADPH Oxidase 3-Derived Reactive Oxygen Species through JNK and P38MAPK Pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence That TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, L.; Liu, W. Cell-Free Synthetic Biology for in Vitro Biosynthesis of Pharmaceutical Natural Products. Synth. Syst. Biotechnol. 2018, 3, 83–89. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the Pleiotropic Role of White Adipose Tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Long, Z.; Zhang, X.; Sun, Q.; Liu, Y.; Liao, N.; Wu, H.; Wang, X.; Hai, C. Evolution of Metabolic Disorder in Rats Fed High Sucrose or High Fat Diet: Focus on Redox State and Mitochondrial Function. Gen. Comp. Endocrinol. 2017, 242, 92–100. [Google Scholar] [CrossRef]
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of Adipokines with Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol. Metab. 2018, 33, 33–43. [Google Scholar] [CrossRef]
- Shabalala, S.C.; Dludla, P.V.; Mabasa, L.; Kappo, A.P.; Basson, A.K.; Pheiffer, C.; Johnson, R. The Effect of Adiponectin in the Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD) and the Potential Role of Polyphenols in the Modulation of Adiponectin Signaling. Biomed. Pharmacother. 2020, 131, 110785. [Google Scholar] [CrossRef]
- Wagner, R.; Eckstein, S.S.; Yamazaki, H.; Gerst, F.; Machann, J.; Jaghutriz, B.A.; Schürmann, A.; Solimena, M.; Singer, S.; Königsrainer, A.; et al. Metabolic Implications of Pancreatic Fat Accumulation. Nat. Rev. Endocrinol. 2021, 18, 43–54. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The Role of Skeletal Muscle Insulin Resistance in the Pathogenesis of the Metabolic Syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I.H.; Newsholme, P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and β-Cell Dysfunction. Oxidative Med. Cell. Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased Oxidative Stress in Obesity and Its Impact on Metabolic Syndrome. J. Clin. Investig. 2017, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Adipose Tissue in Obesity—An Inflammatory Issue. Endocrinology 2005, 146, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef] [Green Version]
- Im, J.A.; Kim, S.H.; Lee, J.W.; Shim, J.Y.; Lee, H.R.; Lee, D.C. Association between Hypoadiponectinemia and Cardiovascular Risk Factors in Nonobese Healthy Adults. Metab. Clin. Exp. 2006, 55, 1546–1550. [Google Scholar] [CrossRef]
- Abenavoli, L.; Peta, V. Role of Adipokines and Cytokines in Non-Alcoholic Fatty Liver Disease. Rev. Recent Clin. Trials 2014, 9, 134–140. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in Nonalcoholic Fatty Liver Disease. Metab. Clin. Exp. 2016, 65, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum Total Adiponectin in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Metab. Clin. Exp. 2011, 60, 313–326. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Jung, U.J.; Choi, M.S. Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, P. Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Mantzoros, C.S. Nonalcoholic Fatty Future Disease. Metab. Clin. Exp. 2016, 65, 1007–1016. [Google Scholar] [CrossRef]
- Maggio, A.B.R.; Mueller, P.; Wacker, J.; Viallon, M.; Belli, D.C.; Beghetti, M.; Farpour-Lambert, N.J.; McLin, V.A. Increased Pancreatic Fat Fraction Is Present in Obese Adolescents with Metabolic Syndrome. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; di Martino, M.; Anania, C.; Andreoli, G.M.; Bezzi, M.; Catalano, C.; Chiesa, C. Pancreatic Fat and β-Cell Function in Overweight/Obese Children with Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2015, 21, 4688–4695. [Google Scholar] [CrossRef] [PubMed]
- Lê, K.A.; Ventura, E.E.; Fisher, J.Q.; Davis, J.N.; Weigensberg, M.J.; Punyanitya, M.; Hu, H.H.; Nayak, K.S.; Goran, M.I. Ethnic Differences in Pancreatic Fat Accumulation and Its Relationship with Other Fat Depots and Inflammatory Markers. Diabetes Care 2011, 34, 485–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.S.; Wong, G.L.H.; Yeung, D.K.W.; Abrigo, J.M.; Kong, A.P.S.; Chan, R.S.M.; Chim, A.M.L.; Shen, J.; Ho, C.S.; Woo, J.; et al. Fatty Pancreas, Insulin Resistance, and β-Cell Function: A Population Study Using Fat-Water Magnetic Resonance Imaging. Am. J. Gastroenterol. 2014, 109, 589–597. [Google Scholar] [CrossRef]
- Heni, M.; Machann, J.; Staiger, H.; Schwenzer, N.F.; Peter, A.; Schick, F.; Claussen, C.D.; Stefan, N.; Häring, H.U.; Fritsche, A. Pancreatic Fat Is Negatively Associated with Insulin Secretion in Individuals with Impaired Fasting Glucose and/or Impaired Glucose Tolerance: A Nuclear Magnetic Resonance Study. Diabetes Metab. Res. Rev. 2010, 26, 200–205. [Google Scholar] [CrossRef]
- Tushuizen, M.E.; Bunck, M.C.; Pouwels, P.J.; Bontemps, S.; van Waesberghe, J.H.T.; Schindhelm, R.K.; Mari, A.; Heine, R.J.; Diamant, M. Pancreatic Fat Content and β-Cell Function in Men with and Without Type 2 Diabetes. Diabetes Care 2007, 30, 2916–2921. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, R.N. Body Composition in Healthy Aging. Ann. N. Y. Acad. Sci. 2000, 904, 437–448. [Google Scholar] [CrossRef]
- Zamboni, M.; Mazzali, G.; Fantin, F.; Rossi, A.; di Francesco, V. Sarcopenic Obesity: A New Category of Obesity in the Elderly. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.M.; Dai Perrard, X.Y.; Perrard, J.L.; Mansoori, A.; Wayne Smith, C.; Wu, H.; Ballantyne, C.M. Attenuated Adipose Tissue and Skeletal Muscle Inflammation in Obese Mice with Combined CD4+ and CD8+ T Cell Deficiency. Atherosclerosis 2014, 233, 419–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strissel, K.J.; DeFuria, J.; Shaul, M.E.; Bennett, G.; Greenberg, A.S.; Obin, M.S. T Cell Recruitment and Th1 Polarization in Adipose Tissue During Diet-Induced Obesity in C57BL/6 Mice. Obesity 2010, 18, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of Adipose Tissue Dendritic Cells Correlated with Obesity-Associated Insulin-Resistance and Inducing Th17 Responses in Mice and Patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, T.; Liu, L.F.; Lamendola, C.; Shen, L.; Morton, J.; Rivas, H.; Winer, D.; Tolentino, L.; Choi, O.; Zhang, H.; et al. T-Cell Profile in Adipose Tissue Is Associated with Insulin Resistance and Systemic Inflammation in Humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2632–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An Introduction to TRP Channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.J.; Sweet, T.B.; Clapham, D.E. International Union of Basic and Clinical Pharmacology. LXXVI. Current Progress in the Mammalian TRP Ion Channel Family. Pharmacol. Rev. 2010, 62, 381–404. [Google Scholar] [CrossRef]
- Nilius, B.; Szallasi, A. Transient Receptor Potential Channels as Drug Targets: From the Science of Basic Research to the Art of Medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef]
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP Channel Drug Discovery: From Target Validation to Clinical Studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef]
- Clapham, D.E.; Runnels, L.W.; Strübing, C. The Trp Ion Channel Family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient Receptor Potential (TRP) Channels. Subcell. Biochem. 2018, 87, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The Capsaicin Receptor: A Heat-Activated Ion Channel in the Pain Pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Minke, B. The History of the Drosophila TRP Channel: The Birth of a New Channel Superfamily. J. Neurogenet. 2010, 24, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benítez-Angeles, M.; Morales-Lázaro, S.L.; Juárez-González, E.; Rosenbaum, T. TRPV1: Structure, Endogenous Agonists, and Mechanisms. Int. J. Mol. Sci. 2020, 21, 3421. [Google Scholar] [CrossRef]
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct Activation of Capsaicin Receptors by Products of Lipoxygenases: Endogenous Capsaicin-like Substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Östman, J.; Bubb, K.J.; Panayiotou, C.; Priestley, J.V.; Baker, M.D.; Ahluwalia, A. 20-Hydroxyeicosatetraenoic Acid (20-HETE) Is a Novel Activator of Transient Receptor Potential Vanilloid 1 (TRPV1) Channel. J. Biol. Chem. 2012, 287, 13868–13876. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, A.M.; Scotland, P.E.; Akopian, A.N.; Hargreaves, K.M. Activation of TRPV1 in the Spinal Cord by Oxidized Linoleic Acid Metabolites Contributes to Inflammatory Hyperalgesia. Proc. Natl. Acad. Sci. USA 2009, 106, 18820–18824. [Google Scholar] [CrossRef] [Green Version]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.H.; Sørgård, M.; di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid Receptors on Sensory Nerves Mediate the Vasodilator Action of Anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef]
- Yu, W.; Liao, Y.; Huang, Y.; Chen, S.Y.; Sun, Y.; Sun, C.; Wu, Y.; Tang, C.; Du, J.; Jin, H. Endogenous Hydrogen Sulfide Enhances Carotid Sinus Baroreceptor Sensitivity by Activating the Transient Receptor Potential Cation Channel Subfamily V Member 1 (TRPV1) Channel. J. Am. Heart Assoc. 2017, 6, e004971. [Google Scholar] [CrossRef]
- DelloStritto, D.J.; Connell, P.J.; Dick, G.M.; Fancher, I.S.; Klarich, B.; Fahmy, J.N.; Kang, P.T.; Chen, Y.R.; Damron, D.S.; Thodeti, C.K.; et al. Differential Regulation of TRPV1 Channels by H2O2: Implications for Diabetic Microvascular Dysfunction. Basic Res. Cardiol. 2016, 111, 21. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, P.; Krishnan, V.; Fettel, K.; Gao, P.; Zhu, Z.; Ren, J.; Thyagarajan, B. TRPV1 Activation Counters Diet-Induced Obesity through Sirtuin-1 Activation and PRDM-16 Deacetylation in Brown Adipose Tissue. Int. J. Obes. 2017, 41, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.L.; Liu, D.Y.; Ma, L.Q.; Luo, Z.D.; Cao, T.B.; Zhong, J.; Yan, Z.C.; Wang, L.J.; Zhao, Z.G.; Zhu, S.J.; et al. Activation of Transient Receptor Potential Vanilloid Type-1 Channel Prevents Adipogenesis and Obesity. Circ. Res. 2007, 100, 1063–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vriens, J.; Janssens, A.; Prenen, J.; Nilius, B.; Wondergem, R. TRPV Channels and Modulation by Hepatocyte Growth Factor/Scatter Factor in Human Hepatoblastoma (HepG2) Cells. Cell Calcium 2004, 36, 19–28. [Google Scholar] [CrossRef]
- Li, L.; Chen, J.; Ni, Y.; Feng, X.; Zhao, Z.; Wang, P.; Sun, J.; Yu, H.; Yan, Z.; Liu, D.; et al. TRPV1 Activation Prevents Nonalcoholic Fatty Liver through UCP2 Upregulation in Mice. Pflug. Arch. Eur. J. Physiol. 2012, 463, 727–732. [Google Scholar] [CrossRef]
- Lv, Z.; Xu, X.; Sun, Z.; Yang, Y.X.; Guo, H.; Li, J.; Sun, K.; Wu, R.; Xu, J.; Jiang, Q.; et al. TRPV1 Alleviates Osteoarthritis by Inhibiting M1 Macrophage Polarization via Ca2+/CaMKII/Nrf2 Signaling Pathway. Cell Death Dis. 2021, 12, 504. [Google Scholar] [CrossRef]
- Macho, A.; Calzado, M.A.; Muñoz-Blanco, J.; Gómez-Díaz, C.; Gajate, C.; Mollinedo, F.; Navas, P.; Muñoz, E. Selective Induction of Apoptosis by Capsaicin in Transformed Cells: The Role of Reactive Oxygen Species and Calcium. Cell Death Differ. 1999, 6, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Samivel, R.; Kim, D.W.; Son, H.R.; Rhee, Y.-H.; Kim, E.H.; Kim, J.H.; Bae, J.-S.; Chung, Y.-J.; Chung, P.-S.; Raz, E.; et al. The Role of TRPV1 in the CD4 + T Cell-Mediated Inflammatory Response of Allergic Rhinitis. Oncotarget 2015, 7, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.F. TRPV1 Activates T Cells. Sci. Signal. 2014, 7, ec301. [Google Scholar] [CrossRef]
- Akiba, Y.; Kato, S.; Katsube, K.I.; Nakamura, M.; Takeuchi, K.; Ishii, H.; Hibi, T. Transient Receptor Potential Vanilloid Subfamily 1 Expressed in Pancreatic Islet β Cells Modulates Insulin Secretion in Rats. Biochem. Biophys. Res. Commun. 2004, 321, 219–225. [Google Scholar] [CrossRef]
- Diaz-Garcia, C.M.; Morales-Lázaro, S.L.; Sánchez-Soto, C.; Velasco, M.; Rosenbaum, T.; Hiriart, M. Role for the TRPV1 Channel in Insulin Secretion from Pancreatic Beta Cells. J. Membr. Biol. 2014, 247, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Armache, J.P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 Ion Channel Suggests Regulatory Mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Appendino, G.; Owsianik, G. The Transient Receptor Potential Channel TRPA1: From Gene to Pathophysiology. Pflug. Arch. Eur. J. Physiol. 2012, 464, 425–458. [Google Scholar] [CrossRef] [PubMed]
- Ro, J.Y.; Lee, J.S.; Zhang, Y. Activation of TRPV1 and TRPA1 Leads to Muscle Nociception and Mechanical Hyperalgesia. Pain 2009, 144, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.S.; Zhong, L.; Hsieh, T.-H.; Abooj, M.; Bishnoi, M.; Hughes, L.; Premkumar, L.S. Expression of Transient Receptor Potential Ankyrin 1 (TRPA1) and Its Role in Insulin Release from Rat Pancreatic Beta Cells. PLoS ONE 2012, 7, e38005. [Google Scholar] [CrossRef]
- Jensen, T.S. New Understanding of Mechanisms of Painful Diabetic Neuropathy: A Path to Prevention and Better Treatment? Scand. J. Pain 2013, 4, 127–128. [Google Scholar] [CrossRef]
- Traverso, N.; Menini, S.; Cosso, L.; Odetti, P.; Albano, E.; Pronzato, M.A.; Marinari, U.M. Immunological Evidence for Increased Oxidative Stress in Diabetic Rats. Diabetologia 1998, 41, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Li, J.; Chen, G.L.; Ge, Y.; Liu, J.; Xie, K.; Peng, X.; Zhou, W.; Zhong, J.; Zhang, Y.; et al. Cryo-EM Structure of TRPC5 at 2.8-Å Resolution Reveals Unique and Conserved Structural Elements Essential for Channel Function. Sci. Adv. 2019, 5, 7935–7959. [Google Scholar] [CrossRef] [Green Version]
- Zimova, L.; Barvikova, K.; Macikova, L.; Vyklicka, L.; Sinica, V.; Barvik, I.; Vlachova, V. Proximal C-Terminus Serves as a Signaling Hub for TRPA1 Channel Regulation via Its Interacting Molecules and Supramolecular Complexes. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Strübing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 Form a Novel Cation Channel in Mammalian Brain. Neuron 2001, 29, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit Composition of Mammalian Transient Receptor Potential Channels in Living Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zholos, A.V. TRPC5. In Mammalian Transient Receptor Potential (TRP) Cation Channels; Handbook of Experimental Pharmacology 222; Springer: Berlin/Heidelberg, Germany, 2014; pp. 129–156. [Google Scholar] [CrossRef]
- Naylor, J.; Al-Shawaf, E.; McKeown, L.; Manna, P.T.; Porter, K.E.; O’Regan, D.; Muraki, K.; Beech, D.J. TRPC5 Channel Sensitivities to Antioxidants and Hydroxylated Stilbenes. J. Biol. Chem. 2011, 286, 5078–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.Z.; Sukumar, P.; Zeng, F.; Li, J.; Jairaman, A.; English, A.; Naylor, J.; Ciurtin, C.; Majeed, Y.; Milligan, C.J.; et al. TRPC Channel Activation by Extracellular Thioredoxin. Nature 2008, 451, 69–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, P.; Sedo, A.; Li, J.; Wilson, L.A.; O’Regan, D.; Lippiat, J.D.; Porter, K.E.; Kearney, M.T.; Ainscough, J.F.X.; Beech, D.J. Constitutively Active TRPC Channels of Adipocytes Confer a Mechanism for Sensing Dietary Fatty Acids and Regulating Adiponectin. Circ. Res. 2012, 111, 191–200. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient Receptor Potential Cation Channels in Disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Susankova, K.; Tousova, K.; Vyklicky, L.; Teisinger, J.; Vlachova, V. Reducing and Oxidizing Agents Sensitize Heat-Activated Vanilloid Receptor (TRPV1) Current. Mol. Pharmacol. 2006, 70, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Keeble, J.E.; Bodkin, J.V.; Liang, L.; Wodarski, R.; Davies, M.; Fernandes, E.S.; Coelho, C.d.F.; Russell, F.; Graepel, R.; Muscara, M.N.; et al. Hydrogen Peroxide Is a Novel Mediator of Inflammatory Hyperalgesia, Acting via Transient Receptor Potential Vanilloid 1-Dependent and Independent Mechanisms. Pain 2009, 141, 135–142. [Google Scholar] [CrossRef]
- Chuang, H.H.; Lin, S. Oxidative Challenges Sensitize the Capsaicin Receptor by Covalent Cysteine Modification. Proc. Natl. Acad. Sci. USA 2009, 106, 20097–20102. [Google Scholar] [CrossRef] [Green Version]
- Ruan, T.; Lin, Y.J.; Hsu, T.H.; Lu, S.H.; Jow, G.M.; Kou, Y.R. Sensitization by Pulmonary Reactive Oxygen Species of Rat Vagal Lung C-Fibers: The Roles of the TRPV1, TRPA1, and P2X Receptors. PLoS ONE 2014, 9, e91763. [Google Scholar] [CrossRef]
- DelloStritto, D.J.; Sinharoy, P.; Connell, P.J.; Fahmy, J.N.; Cappelli, H.C.; Thodeti, C.K.; Geldenhuys, W.J.; Damron, D.S.; Bratz, I.N. 4-Hydroxynonenal Dependent Alteration of TRPV1-Mediated Coronary Microvascular Signaling. Free Radic. Biol. Med. 2016, 101, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Zhang, L.; Westlund, K.N. Reactive Oxygen Species Mediate TNFR1 Increase after TRPV1 Activation in Mouse DRG Neurons. Mol. Pain 2009, 5, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, E.S.; Liang, L.; Smillie, S.-J.; Kaiser, F.; Purcell, R.; Rivett, D.W.; Alam, S.; Howat, S.; Collins, H.; Thompson, S.J.; et al. TRPV1 Deletion Enhances Local Inflammation and Accelerates the Onset of Systemic Inflammatory Response Syndrome. J. Immunol. 2012, 188, 5741–5751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevisani, M.; Siemens, J.; Materazzi, S.; Bautista, D.M.; Nassini, R.; Campi, B.; Imamachi, N.; Andrè, E.; Patacchini, R.; Cottrell, G.S.; et al. 4-Hydroxynonenal, an Endogenous Aldehyde, Causes Pain and Neurogenic Inflammation through Activation of the Irritant Receptor TRPA1. Proc. Natl. Acad. Sci. USA 2007, 104, 13519–13524. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Mizuno, Y.; Kozai, D.; Yamamoto, S.; Kiyonaka, S.; Shibata, T.; Uchida, K.; Mori, Y. Molecular Characterization of TRPA1 Channel Activation by Cysteine-Reactive Inflammatory Mediators. Channels 2008, 2, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Clark, T.E. Role of Reactive Oxygen Species and TRP Channels in the Cough Reflex. Cell Calcium 2016, 60, 155. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.A.; Gentry, C.; Moss, S.; Bevan, S. Transient Receptor Potential A1 Is a Sensory Receptor for Multiple Products of Oxidative Stress. J. Neurosci. 2008, 28, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, S.; Yuan, S.Y.; Brookes, S.J.H.; Spencer, N.J.; Zagorodnyuk, V.P. Hydrogen Peroxide Preferentially Activates Capsaicin-Sensitive High Threshold Afferents via TRPA1 Channels in the Guinea Pig Bladder. Br. J. Pharmacol. 2017, 174, 126–138. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, L.J.; Xiao, B.; Kwan, K.Y.; Petrus, M.J.; Dubin, A.E.; Hwang, S.W.; Cravatt, B.; Corey, D.P.; Patapoutian, A. An Ion Channel Essential for Sensing Chemical Damage. J. Neurosci. 2007, 27, 11412–11415. [Google Scholar] [CrossRef]
- Bessac, B.F.; Sivula, M.; von Hehn, C.A.; Escalera, J.; Cohn, L.; Jordt, S.E. TRPA1 Is a Major Oxidant Sensor in Murine Airway Sensory Neurons. J. Clin. Investig. 2008, 118, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Vascular Biology: Localized TRPA1 Channel Ca2+ Signals Stimulated by Reactive Oxygen Species Promote Cerebral Artery Dilation. Sci. Signal. 2015, 8, ra2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubdool, A.A.; Kodji, X.; Abdul-Kader, N.; Heads, R.; Fernandes, E.S.; Bevan, S.; Brain, S.D. TRPA1 Activation Leads to Neurogenic Vasodilatation: Involvement of Reactive Oxygen Nitrogen Species in Addition to CGRP and NO. Br. J. Pharmacol. 2016, 173, 2419–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunggip, C.; Shimoda, K.; Oda, S.; Tanaka, T.; Nishiyama, K.; Mangmool, S.; Nishimura, A.; Numaga-Tomita, T.; Nishida, M. TRPC5-ENOS Axis Negatively Regulates ATP-Induced Cardiomyocyte Hypertrophy. Front. Pharmacol. 2018, 9, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Shimizu, S. Significance of TRP Channels in Oxidative Stress. Eur. J. Pharmacol. 2016, 793, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Dublin, A.E.; Petrus, M.J.; Patapoutian, A. TRPV1 and TRPA1 Mediate Peripheral Nitric Oxide-Induced Nociception in Mice. PLoS ONE 2009, 4, e7596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duo, L.; Wu, T.; Ke, Z.; Hu, L.; Wang, C.; Teng, G.; Zhang, W.; Wang, W.; Ge, Q.; Yang, Y.; et al. Gain of Function of Ion Channel TRPV1 Exacerbates Experimental Colitis by Promoting Dendritic Cell Activation. Mol. Ther. Nucleic Acids 2020, 22, 924–936. [Google Scholar] [CrossRef]
- Santos Pereira, D.M.; Teixeira, S.A.; Murillo, O.; Machado Peixoto, E.P.; Araújo, M.C.; Fialho Sousa, N.C.; Monteiro-Neto, V.; Calixto, J.B.; Cunha, T.M.; Farias Marinho, C.R.; et al. TRPV1 Contributes to Cerebral Malaria Severity and Mortality by Regulating Brain Inflammation. Oxidative Med. Cell. Longev. 2019, 2019, 9451671. [Google Scholar] [CrossRef] [Green Version]
- Bok, E.; Chung, Y.C.; Kim, K.S.; Baik, H.H.; Shin, W.H.; Jin, B.K. Modulation of M1/M2 Polarization by Capsaicin Contributes to the Survival of Dopaminergic Neurons in the Lipopolysaccharide-Lesioned Substantia Nigra in Vivo. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Von Banchet, G.S.; Richter, J.; Hückel, M.; Rose, C.; Bräuer, R.; Schaible, H.G. Fibroblast-like Synovial Cells from Normal and Inflamed Knee Joints Differently Affect the Expression of Pain-Related Receptors in Sensory Neurones: A Co-Culture Study. Arthritis Res. Ther. 2007, 9, R6. [Google Scholar] [CrossRef] [Green Version]
- Marshall, N.J.; Liang, L.; Bodkin, J.; Dessapt-Baradez, C.; Nandi, M.; Collot-Teixeira, S.; Smillie, S.J.; Lalgi, K.; Fernandes, E.S.; Gnudi, L.; et al. A Role for TRPV1 in Influencing the Onset of Cardiovascular Disease in Obesity. Hypertension 2013, 61, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Ye, L.; Zhang, Q.; Wu, F.; Wang, L. The Role of TRPV1 Channels in Atherosclerosis. Channels 2020, 14, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, A.; Wakano, C.; Koblan-Huberson, M.; Adra, C.N.; Fleig, A.; Turner, H. TRPA1 Is a Substrate for De-Ubiquitination by the Tumor Suppressor CYLD. Cell. Signal. 2006, 18, 1584–1594. [Google Scholar] [CrossRef]
- Bertin, S.; Aoki-Nonaka, Y.; Lee, J.; de Jong, P.R.; Kim, P.; Han, T.; Yu, T.; To, K.; Takahashi, N.; Boland, B.S.; et al. The TRPA1 Ion Channel Is Expressed in CD4+ T Cells and Restrains T-Cell-Mediated Colitis through Inhibition of TRPV1. Gut 2017, 66, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Pereira, I.C.d.P.; Mendes, S.J.F.; Pereira, D.M.S.; Muniz, T.F.; Colares, V.L.P.; Monteiro, C.R.A.V.; Martins, M.M.R.d.S.; Grisotto, M.A.G.; Monteiro-Neto, V.; Monteiro, S.G.; et al. Transient Receptor Potential Ankyrin 1 Channel Expression on Peripheral Blood Leukocytes from Rheumatoid Arthritic Patients and Correlation with Pain and Disability. Front. Pharmacol. 2017, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, S.S.; Majhi, R.K.; Tiwari, A.; Acharya, T.; Kumar, P.S.; Saha, S.; Kumar, A.; Goswami, C.; Chattopadhyay, S. Transient Receptor Potential Ankyrin1 Channel Is Endogenously Expressed in T Cells and Is Involved in Immune Functions. Biosci. Rep. 2019, 39, BSR20191437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Han, X.; He, L.; Tang, F.; Huang, R.; Lin, Z.; Li, S.; Deng, S.; Xu, J.; Huang, H.; et al. Transient Receptor Potential Ankyrin 1 Contributes to the ATP-Elicited Oxidative Stress and Inflammation in THP-1-Derived Macrophage. Mol. Cell. Biochem. 2020, 473, 179–192. [Google Scholar] [CrossRef]
- Tian, C.; Huang, R.; Tang, F.; Lin, Z.; Cheng, N.; Han, X.; Li, S.; Zhou, P.; Deng, S.; Huang, H.; et al. Transient Receptor Potential Ankyrin 1 Contributes to Lysophosphatidylcholine-Induced Intracellular Calcium Regulation and THP-1-Derived Macrophage Activation. J. Membr. Biol. 2020, 253, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Kobayashi, T.; Kamata, K. Role of Lysophosphatidylcholine (LPC) in Atherosclerosis. Curr. Med. Chem. 2007, 14, 3209–3220. [Google Scholar] [CrossRef] [Green Version]
- Stachon, P.; Geis, S.; Peikert, A.; Heidenreich, A.; Michel, N.A.; Ünal, F.; Hoppe, N.; Dufner, B.; Schulte, L.; Marchini, T.; et al. Extracellular ATP Induces Vascular Inflammation and Atherosclerosis via Purinergic Receptor y 2 in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1577–1586. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.V.; Li, Y.; Liu, X.; Xia, S.; Shi, P.; Li, L.; Chen, Z.; Yin, C.; Eriguchi, M.; Chen, Y.; et al. ATP Release Drives Heightened Immune Responses Associated with Hypertension. Sci. Immunol. 2019, 4, eaau6426. [Google Scholar] [CrossRef]
- Zhao, J.F.; Shyue, S.K.; Kou, Y.R.; Lu, T.M.; Lee, T.S. Transient Receptor Potential Ankyrin 1 Channel Involved in Atherosclerosis and Macrophage-Foam Cell Formation. Int. J. Biol. Sci. 2016, 12, 812–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lu, Z.-H.; Gabius, H.-J.; Rohowsky-Kochan, C.; Ledeen, R.W.; Wu, G. Cross-Linking of GM1 Ganglioside by Galectin-1 Mediates Regulatory T Cell Activity Involving TRPC5 Channel Activation: Possible Role in Suppressing Experimental Autoimmune Encephalomyelitis. J. Immunol. 2009, 182, 4036–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Guo, G.; Qi, Y.; Xiong, Y.; Ma, X.; Wu, N.; Dong, C.; Yang, C. Inhibition of Canonical Transient Receptor Potential 5 Channels Polarizes Macrophages to an M1 Phenotype. Pharmacology 2020, 105, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.S.; Mendes, S.J.F.; Alawi, K.; Thakore, P.; Aubdool, A.; Sousa, N.C.F.; da Silva, J.F.R.; Castro, J.A.; Pereira, I.C.P.; Silva, L.C.N.; et al. Transient Receptor Potential Canonical Channels 4 and 5 Mediate Escherichia Coli-Derived Thioredoxin Effects in Lipopolysaccharide-Injected Mice. Oxidative Med. Cell. Longev. 2018, 2018, 4904696. [Google Scholar] [CrossRef] [Green Version]
- Gunawardena, D.; Raju, R.; Münch, G. Hydrogen Peroxide Mediates Pro-Inflammatory Cell-to-Cell Signaling: A New Therapeutic Target for Inflammation? Neural Regen. Res. 2019, 14, 1430–1437. [Google Scholar] [CrossRef]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial Respiratory Chain-Dependent Generation of Superoxide Anion and Its Release into the Intermembrane Space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef]
- Nakao, N.; Kurokawa, T.; Nonami, T.; Tumurkhuu, G.; Koide, N.; Yokochi, T. Hydrogen Peroxide Induces the Production of Tumor Necrosis Factor-Alpha in RAW 264.7 Macrophage Cells via Activation of P38 and Stress-Activated Protein Kinase. Innate Immun. 2008, 14, 190–196. [Google Scholar] [CrossRef]
- Hancock, J.T. The Role of Redox Mechanisms in Cell Signalling. Mol. Biotechnol. 2009, 43, 162–166. [Google Scholar] [CrossRef]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A Tissue-Scale Gradient of Hydrogen Peroxide Mediates Rapid Wound Detection in Zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Kumagai, T.; Matsukawa, N.; Kaneko, Y.; Kusumi, Y.; Mitsumata, M.; Uchida, K. A Lipid Peroxidation-Derived Inflammatory Mediator: Identification of 4-Hydroxy-2-Nonenal as a Potential Inducer of Cyclooxygenase-2 in Macrophages. J. Biol. Chem. 2004, 279, 48389–48396. [Google Scholar] [CrossRef] [Green Version]
- Ruef, J.; Moser, M.; Bode, C.; Kübler, W.; Runge, M.S. 4-Hydroxynonenal Induces Apoptosis, NF-KappaB-Activation and Formation of 8-Isoprostane in Vascular Smooth Muscle Cells. Basic Res. Cardiol. 2001, 96, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, C.E.; Seo, K.W.; Kim, C.D. HNE-Induced 5-LO Expression Is Regulated by NF-ΚB/ERK and Sp1/P38 MAPK Pathways via EGF Receptor in Murine Macrophages. Cardiovasc. Res. 2010, 88, 352–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C.; Kozak, K.R.; Marnett, L.J. IkappaB Kinase, a Molecular Target for Inhibition by 4-Hydroxy-2-Nonenal. J. Biol. Chem. 2001, 276, 18223–18228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, S.; Fischer, C.; Baumgartner, B.; Haas, M.; Kreusel, U.; Loidl, G.; Hayn, M.; Ziegler-Heitbrock, H.W.L.; Neumeier, D.; Brand, K. 4-Hydroxynonenal Prevents NF-KappaB Activation and Tumor Necrosis Factor Expression by Inhibiting IkappaB Phosphorylation and Subsequent Proteolysis. J. Biol. Chem. 1999, 274, 11611–11618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hadri, K.; Dler Faieeq Darweesh, M.; Couchie, D.; Jguirim-Souissi, I.; Genze, F.; Diderot, V.; Syrovets, T.; Lunov, O.; Simmet, T.; Rouis, M. Thioredoxin-1 Promotes Anti-Inflammatory Macrophages of the M2 Phenotype and Antagonizes Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1445–1452. [Google Scholar] [CrossRef] [Green Version]
- Canesi, F.; Mateo, V.; Couchie, D.; Karabina, S.; Nègre-Salvayre, A.; Rouis, M.; el Hadri, K. A Thioredoxin-Mimetic Peptide Exerts Potent Anti-Inflammatory, Antioxidant, and Atheroprotective Effects in ApoE2.Ki Mice Fed High Fat Diet. Cardiovasc. Res. 2019, 115, 292–301. [Google Scholar] [CrossRef]
- Bishnoi, M.; Kondepudi, K.K.; Gupta, A.; Karmase, A.; Boparai, R.K. Expression of Multiple Transient Receptor Potential Channel Genes in Murine 3T3-L1 Cell Lines and Adipose Tissue. Pharmacol. Rep. 2013, 65, 751–755. [Google Scholar] [CrossRef] [Green Version]
- Baskaran, P.; Krishnan, V.; Ren, J.; Thyagarajan, B. Capsaicin Induces Browning of White Adipose Tissue and Counters Obesity by Activating TRPV1 Channel-Dependent Mechanisms. Br. J. Pharmacol. 2016, 173, 2369–2389. [Google Scholar] [CrossRef]
- Xin, H.; Tanaka, H.; Yamaguchi, M.; Takemori, S.; Nakamura, A.; Kohama, K. Vanilloid Receptor Expressed in the Sarcoplasmic Reticulum of Rat Skeletal Muscle. Biochem. Biophys. Res. Commun. 2005, 332, 756–762. [Google Scholar] [CrossRef]
- Cavuoto, P.; McAinch, A.J.; Hatzinikolas, G.; Janovská, A.; Game, P.; Wittert, G.A. The Expression of Receptors for Endocannabinoids in Human and Rodent Skeletal Muscle. Biochem. Biophys. Res. Commun. 2007, 364, 105–110. [Google Scholar] [CrossRef]
- Page, A.J.; Hatzinikolas, G.; Vincent, A.D.; Cavuoto, P.; Wittert, G.A. The TRPV1 Channel Regulates Glucose Metabolism. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E667–E676. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Lee, D.K.; Liu, S.M.; Chua, S.C.; Schwartz, G.J.; Jo, Y.H. Activation of Temperature-Sensitive TRPV1-like Receptors in ARC POMC Neurons Reduces Food Intake. PLoS Biol. 2018, 16, e2004399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Tu, Y.; Li, B.; Zhang, L.; Feng, L.; Wang, L.; Zhang, L.; Zhou, H.; Hua, X.; Ma, X. Roux-En-Y Gastric Bypass Enhances Insulin Secretion in Type 2 Diabetes via FXR-Mediated TRPA1 Expression. Mol. Metab. 2019, 29, 1–11. [Google Scholar] [CrossRef]
- Osterloh, M.; Böhm, M.; Kalbe, B.; Osterloh, S.; Hatt, H. Identification and Functional Characterization of TRPA1 in Human Myoblasts. Pflug. Arch. Eur. J. Physiol. 2016, 468, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Gao, Y.; Yao, T.; Deng, Z.; Sohn, J.W.; Sun, J.; Huang, Y.; Kong, X.; Yu, K.-J.; Wang, R.-T.; Chen, H.; et al. TrpC5 Mediates Acute Leptin and Serotonin Effects via Pomc Neurons. Cell Rep. 2017, 18, 583–592. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Gao, Y.; Lieu, L.; Afrin, S.; Cao, J.; Michael, N.J.; Dong, Y.; Sun, J.; Guo, H.; Williams, K.W. Direct and Indirect Effects of Liraglutide on Hypothalamic POMC and NPY/AgRP Neurons—Implications for Energy Balance and Glucose Control. Mol. Metab. 2019, 28, 120–134. [Google Scholar] [CrossRef]
- Dong, Y.; Carty, J.; Goldstein, N.; He, Z.; Hwang, E.; Chau, D.; Wallace, B.; Kabahizi, A.; Lieu, L.; Peng, Y.; et al. Time and Metabolic State-Dependent Effects of GLP-1R Agonists on NPY/AgRP and POMC Neuronal Activity in Vivo. Mol. Metab. 2021, 54, 101352. [Google Scholar] [CrossRef]
- Qian, F.; Huang, P.; Ma, L.; Kuznetsov, A.; Tamarina, N.; Philipson, L.H. TRP GenesCandidates for Nonselective Cation Channels and Store-Operated Channels in Insulin-Secreting Cells. Diabetes 2002, 51, S183–S189. [Google Scholar] [CrossRef] [Green Version]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 Activation by Cyclic ADP-Ribose at Body Temperature Is Involved in Insulin Secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef]
- Liu, D.; Zhu, Z.; Tepel, M. The Role of Transient Receptor Potential Channels in Metabolic Syndrome. Hypertens. Res. 2008, 31, 1989–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, R.; Chan, Y.; Afifiyan, F.N.; Liu, X.J.; Wan, X.; Yantha, J.; Tsui, H.; Tang, L.; Tsai, S.; Santamaria, P.; et al. TRPV1+ Sensory Neurons Control Beta Cell Stress and Islet Inflammation in Autoimmune Diabetes. Cell 2006, 127, 1123–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera, C.E.; Huising, M.O.; Follett, P.; Leblanc, M.; Halloran, J.; van Andel, R.; de Magalhaes Filho, C.D.; Merkwirth, C.; Dillin, A. TRPV1 Pain Receptors Regulate Longevity and Metabolism by Neuropeptide Signaling. Cell 2014, 157, 1023–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gram, D.X.; Hansen, A.J.; Wilken, M.; Elm, T.; Svendsen, O.; Carr, R.D.; Ahrén, B.; Brand, C.L. Plasma Calcitonin Gene-Related Peptide Is Increased Prior to Obesity, and Sensory Nerve Desensitization by Capsaicin Improves Oral Glucose Tolerance in Obese Zucker Rats. Eur. J. Endocrinol. 2005, 153, 963–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Shimaya, A.; Kiso, T.; Kuramochi, T.; Shimokawa, T.; Shibasaki, M. Enhanced Insulin Secretion and Sensitization in Diabetic Mice on Chronic Treatment with a Transient Receptor Potential Vanilloid 1 Antagonist. Life Sci. 2011, 88, 559–563. [Google Scholar] [CrossRef]
- Lee, E.; Jung, D.Y.; Kim, J.H.; Patel, P.R.; Hu, X.; Lee, Y.; Azuma, Y.; Wang, H.F.; Tsitsilianos, N.; Shafiq, U.; et al. Transient Receptor Potential Vanilloid Type-1 Channel Regulates Diet-Induced Obesity, Insulin Resistance, and Leptin Resistance. FASEB J. 2015, 29, 3182–3192. [Google Scholar] [CrossRef] [Green Version]
- Ávila, D.L.; Nunes, N.A.M.; Almeida, P.H.R.F.; Gomes, J.A.S.; Rosa, C.O.B.; Alvarez-Leite, J.I. Signaling Targets Related to Antiobesity Effects of Capsaicin: A Scoping Review. Adv. Nutr. 2021, 12, 2232–2243. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Guo, Z.; Li, T.P.; Sun, T. Dietary Capsaicin Normalizes CGRP Peptidergic DRG Neurons in Experimental Diabetic Peripheral Neuropathy. Sci. Rep. 2021, 11, 1704. [Google Scholar] [CrossRef]
- Anand, P.; Murali, K.Y.; Tandon, V.; Murthy, P.S.; Chandra, R. Insulinotropic Effect of Cinnamaldehyde on Transcriptional Regulation of Pyruvate Kinase, Phosphoenolpyruvate Carboxykinase, and GLUT4 Translocation in Experimental Diabetic Rats. Chem. Biol. Interact. 2010, 186, 72–81. [Google Scholar] [CrossRef]
- Chepurny, O.G.; Leech, C.A.; Tomanik, M.; Dipoto, M.C.; Li, H.; Han, X.; Meng, Q.; Cooney, R.N.; Wu, J.; Holz, G.G. Synthetic Small Molecule GLP-1 Secretagogues Prepared by Means of a Three-Component Indole Annulation Strategy. Sci. Rep. 2016, 6, 28934. [Google Scholar] [CrossRef]
- Kim, M.J.; Son, H.J.; Song, S.H.; Jung, M.; Kim, Y.; Rhyu, M.R. The TRPA1 Agonist, Methyl Syringate Suppresses Food Intake and Gastric Emptying. PLoS ONE 2013, 8, e71603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Chen, X.; Cerne, R.; Syed, S.K.; Ficorilli, J.V.; Cabrera, O.; Obukhov, A.G.; Efanov, A.M. Catechol Estrogens Stimulate Insulin Secretion in Pancreatic β-Cells via Activation of the Transient Receptor Potential A1 (TRPA1) Channel. J. Biol. Chem. 2019, 294, 2935–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, D.A.; Filipovic, M.R.; Gentry, C.; Eberhardt, M.; Vastani, N.; Leffler, A.; Reeh, P.; Bevan, S. Streptozotocin Stimulates the Ion Channel TRPA1 Directly: Involvement of Peroxynitrite. J. Biol. Chem. 2015, 290, 15185–15196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaruzza, F.; Johnson, C.; Leggit, A.; Grady, E.; Katrin Schenk, A.; Cevikbas, F.; Cedron, W.; Bondada, S.; Kirkwood, R.; Malone, B.; et al. Transient Receptor Potential Ankyrin 1 Mediates Chronic Pancreatitis Pain in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1002–G1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Lee, H.; Im, S.W.; Jung, C.H.; Ha, T.Y. Allyl Isothiocyanate Ameliorates Insulin Resistance through the Regulation of Mitochondrial Function. J. Nutr. Biochem. 2014, 25, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulère, L.; Lagarde, M.; Soulage, C.O. The Lipid Peroxidation By-Product 4-Hydroxy-2-Nonenal (4-HNE) Induces Insulin Resistance in Skeletal Muscle through Both Carbonyl and Oxidative Stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, X.M.; Zhang, Y.B.; Huang, X.; Chi, M.H. Association of 4-Hydroxynonenal with Classical Adipokines and Insulin Resistance in a Chinese Non-Diabetic Obese Population. Nutr. Hosp. 2017, 34, 363–368. [Google Scholar] [CrossRef]
- Marabita, F.; Islam, M.S. Expression of Transient Receptor Potential Channels in the Purified Human Pancreatic β-Cells. Pancreas 2017, 46, 97–101. [Google Scholar] [CrossRef]
- Fels, B.; Nielsen, N.; Schwab, A. Role of TRPC1 Channels in Pressure-Mediated Activation of Murine Pancreatic Stellate Cells. Eur. Biophys. J. EBJ 2016, 45, 657–670. [Google Scholar] [CrossRef]
- Krout, D.; Schaar, A.; Sun, Y.; Sukumaran, P.; Roemmich, J.N.; Singh, B.B.; Claycombe-Larson, K.J. The TRPC1 Ca2+-Permeable Channel Inhibits Exercise-Induced Protection against High-Fat Diet-Induced Obesity and Type II Diabetes. J. Biol. Chem. 2017, 292, 20799–20807. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, W.; Cui, W.; Shi, B.; Wang, H. PKCα Promotes Insulin Secretion via TRPC1 Phosphorylation in INS-1E Cells. Biosci. Biotechnol. Biochem. 2019, 83, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ning, K.; Sun, X.; Zhang, C.; Jin, L.-F.; Hua, D. Glycolysis Is Essential for Chemoresistance Induced by Transient Receptor Potential Channel C5 in Colorectal Cancer. BMC Cancer 2018, 18, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dattilo, M.; Penington, N.J.; Williams, K. Inhibition of TRPC5 Channels by Intracellular ATP. Mol. Pharmacol. 2008, 73, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered Adipose Tissue and Adipocyte Function in the Pathogenesis of Metabolic Syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q.; et al. Brown and Beige Adipose Tissue: A Novel Therapeutic Strategy for Obesity and Type 2 Diabetes Mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Tamura, Y.; Inayoshi, K.; Narukawa, M.; Kobata, K.; Chiba, H.; Muraki, E.; Tsunoda, N.; Watanabe, T. TRPV1 Agonist Monoacylglycerol Increases UCP1 Content in Brown Adipose Tissue and Suppresses Accumulation of Visceral Fat in Mice Fed a High-Fat and High-Sucrose Diet. Biosci. Biotechnol. Biochem. 2011, 75, 904–909. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, F.; Piper, M.; Ho, L.L.; Huang, T.L.; Gupta, A.; Streets, A.; Lynes, M.D.; Tseng, Y.H. Vascular Smooth Muscle-Derived Trpv1+ Progenitors Are a Source of Cold-Induced Thermogenic Adipocytes. Nat. Metab. 2021, 3, 485–495. [Google Scholar] [CrossRef]
- Motter, A.L.; Ahern, G.P. TRPV1-Null Mice Are Protected from Diet-Induced Obesity. FEBS Lett. 2008, 582, 2257–2262. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.O.; Moritani, T. Alterations of Autonomic Nervous Activity and Energy Metabolism by Capsaicin Ingestion during Aerobic Exercise in Healthy Men. J. Nutr. Sci. Vitaminol. 2007, 53, 124–132. [Google Scholar] [CrossRef] [Green Version]
- Lieder, B.; Zaunschirm, M.; Holik, A.K.; Ley, J.P.; Hans, J.; Krammer, G.E.; Somoza, V. The Alkamide Trans-Pellitorine Targets PPARγ via TRPV1 and TRPA1 to Reduce Lipid Accumulation in Developing 3T3-L1 Adipocytes. Front. Pharmacol. 2017, 8, 316. [Google Scholar] [CrossRef]
- Hoi, J.K.; Lieder, B.; Liebisch, B.; Czech, C.; Hans, J.; Ley, J.P.; Somoza, V. TRPA1 Agonist Cinnamaldehyde Decreases Adipogenesis in 3T3-L1 Cells More Potently than the Non-Agonist Structural Analog Cinnamyl Isobutyrate. ACS Omega 2020, 5, 33305–33313. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Emont, M.P.; Jun, H.; Qiao, X.; Liao, J.; Kim, D.-i.; Wu, J. Cinnamaldehyde Induces Fat Cell-Autonomous Thermogenesis and Metabolic Reprogramming. Metab. Clin. Exp. 2017, 77, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Terada, Y. Food Compounds Activating Thermosensitive TRP Channels in Asian Herbal and Medicinal Foods. J. Nutr. Sci. Vitaminol. 2015, 61, S86–S88. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yuan, H.D.; Kim, D.Y.; Quan, H.Y.; Chung, S.H. Cinnamaldehyde Prevents Adipocyte Differentiation and Adipogenesis via Regulation of Peroxisome Proliferator-Activated Receptor-γ (PPARγ) and AMP-Activated Protein Kinase (AMPK) Pathways. J. Agric. Food Chem. 2011, 59, 3666–3673. [Google Scholar] [CrossRef]
- Elrayess, M.A.; Almuraikhy, S.; Kafienah, W.; Al-Menhali, A.; Al-Khelaifi, F.; Bashah, M.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Alsayrafi, M.; et al. 4-Hydroxynonenal Causes Impairment of Human Subcutaneous Adipogenesis and Induction of Adipocyte Insulin Resistance. Free Radic. Biol. Med. 2017, 104, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Dou, X.; Gu, D.; Shen, C.; Yao, T.; Nguyen, V.; Braunschweig, C.; Song, Z. 4-Hydroxynonenal Differentially Regulates Adiponectin Gene Expression and Secretion via Activating PPARγ and Accelerating Ubiquitin-Proteasome Degradation. Mol. Cell. Endocrinol. 2012, 349, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.M.; Guo, L.; Huang, X.; Li, Q.M.; Chi, M.H. 4-Hydroxynonenal Regulates TNF-α Gene Transcription Indirectly via ETS1 and MicroRNA-29b in Human Adipocytes Induced from Adipose Tissue-Derived Stromal Cells. Anat. Rec. 2016, 299, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Satapati, S.; Kucejova, B.; Duarte, J.A.G.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Arya Nair, L.; Livingston, K.; Fu, X.; et al. Mitochondrial Metabolism Mediates Oxidative Stress and Inflammation in Fatty Liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Luo, H.; Jiang, Y.; Chen, P.; Hu, J.; Luo, H.; Jiang, Y.; Chen, P. Dietary Capsaicin and Antibiotics Act Synergistically to Reduce Non-Alcoholic Fatty Liver Disease Induced by High Fat Diet in Mice. Oncotarget 2017, 8, 38161–38175. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Pu, Y.; Wang, P.; Chen, S.; Zhao, Y.; Liu, C.; Shang, Q.; Zhu, Z.; Liu, D. TRPV1-Mediated UCP2 Upregulation Ameliorates Hyperglycemia-Induced Endothelial Dysfunction. Cardiovasc. Diabetol. 2013, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.F.; Ching, L.C.; Kou, Y.R.; Lin, S.J.; Wei, J.; Shyue, S.K.; Lee, T.S. Activation of TRPV1 Prevents OxLDL-Induced Lipid Accumulation and TNF-α-Induced Inflammation in Macrophages: Role of Liver X Receptor α. Mediat. Inflamm. 2013, 2013, 925171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Li, L.; Wang, F.; Chen, J.; Zhao, Y.; Wang, P.; Nilius, B.; Liu, D.; Zhu, Z. Dietary Capsaicin Prevents Nonalcoholic Fatty Liver Disease through Transient Receptor Potential Vanilloid 1-Mediated Peroxisome Proliferator-Activated Receptor δ Activation. Pflug. Arch. Eur. J. Physiol. 2013, 465, 1303–1316. [Google Scholar] [CrossRef]
- Baskaran, P.; Nazminia, K.; Frantz, J.; O’Neal, J.; Thyagarajan, B. Mice Lacking Endogenous TRPV1 Express Reduced Levels of Thermogenic Proteins and Are Susceptible to Diet-Induced Obesity and Metabolic Dysfunction. FEBS Lett. 2021, 595, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Beier, J.I.; Arteel, G.E.; Ramsden, C.E.; Feldstein, A.E.; McClain, C.J.; Kirpich, I.A. Transient Receptor Potential Vanilloid 1 Gene Deficiency Ameliorates Hepatic Injury in a Mouse Model of Chronic Binge Alcohol-Induced Alcoholic Liver Disease. Am. J. Pathol. 2015, 185, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Abdelmageed, M.E.; Shehatou, G.S.; Abdelsalam, R.A.; Suddek, G.M.; Salem, H.A. Cinnamaldehyde Ameliorates STZ-Induced Rat Diabetes through Modulation of IRS1/PI3K/AKT2 Pathway and AGEs/RAGE Interaction. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 243–258. [Google Scholar] [CrossRef]
- Hosni, A.A.; Abdel-Moneim, A.A.; Abdel-Reheim, E.S.; Mohamed, S.M.; Helmy, H. Cinnamaldehyde Potentially Attenuates Gestational Hyperglycemia in Rats through Modulation of PPARγ, Proinflammatory Cytokines and Oxidative Stress. Biomed. Pharmacother. 2017, 88, 52–60. [Google Scholar] [CrossRef]
- Badr, H.; Kozai, D.; Sakaguchi, R.; Numata, T.; Mori, Y. Different Contribution of Redox-Sensitive Transient Receptor Potential Channels to Acetaminophen-Induced Death of Human Hepatoma Cell Line. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Alawi, K.M.; Tandio, D.; Xu, J.; Thakore, P.; Papacleovoulou, G.; Fernandes, E.S.; Legido-Quigley, C.; Williamson, C.; Brain, S.D. Transient Receptor Potential Canonical 5 Channels Plays an Essential Role in Hepatic Dyslipidemia Associated with Cholestasis. Sci. Rep. 2017, 7, 2338. [Google Scholar] [CrossRef] [Green Version]
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The Metabolic Syndrome: Role of Skeletal Muscle Metabolism. Ann. Med. 2006, 38, 389–402. [Google Scholar] [CrossRef]
- Kim, G.; Kim, J.H. Impact of Skeletal Muscle Mass on Metabolic Health. Endocrinol. Metab. 2020, 35, 1–6. [Google Scholar] [CrossRef]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.P.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted Disruption of the Glucose Transporter 4 Selectively in Muscle Causes Insulin Resistance and Glucose Intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, S.; Raschke, S.; Knebel, B.; Scheler, M.; Irmler, M.; Passlack, W.; Muller, S.; Hanisch, F.G.; Franz, T.; Li, X.; et al. Secretome Profiling of Primary Human Skeletal Muscle Cells. Biochim. Biophys. Acta 2014, 1844, 1011–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raschke, S.; Eckardt, K.; Bjørklund Holven, K.; Jensen, J.; Eckel, J. Identification and Validation of Novel Contraction-Regulated Myokines Released from Primary Human Skeletal Muscle Cells. PLoS ONE 2013, 8, e62008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, Y.K.; Kim, S.S.; Kim, J.H.; Kim, H.J.; Kim, H.J.; Park, J.J.; Cho, Y.S.; Joung, S.H.; Kim, J.R.; Kim, B.H.; et al. Combined Aerobic and Resistance Exercise Training Reduces Circulating Apolipoprotein J Levels and Improves Insulin Resistance in Postmenopausal Diabetic Women. Diabetes Metab. J. 2020, 44, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Ma, L.; Zhao, Z.; He, H.; Yang, D.; Feng, X.; Ma, S.; Chen, X.; Zhu, T.; Cao, T.; et al. TRPV1 Activation Improves Exercise Endurance and Energy Metabolism through PGC-1α Upregulation in Mice. Cell Res. 2011, 22, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Ferdowsi, P.V.; Ahuja, K.D.K.; Beckett, J.M.; Myers, S. TRPV1 Activation by Capsaicin Mediates Glucose Oxidation and ATP Production Independent of Insulin Signalling in Mouse Skeletal Muscle Cells. Cells 2021, 10, 1560. [Google Scholar] [CrossRef]
- Pilegaard, H.; Saltin, B.; Neufer, D.P. Exercise Induces Transient Transcriptional Activation of the PGC-1α Gene in Human Skeletal Muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef]
- Zanou, N.; Shapovalov, G.; Louis, M.; Tajeddine, N.; Gallo, C.; van Schoor, M.; Anguish, I.; Cao, M.L.; Schakman, O.; Dietrich, A.; et al. Role of TRPC1 Channel in Skeletal Muscle Function. Am. J. Physiol. Cell Physiol. 2010, 298, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Sabourin, J.; Saüc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 Channels Functionally Interact with STIM1L to Promote Myogenesis and Maintain Fast Repetitive Ca2 + Release in Human Myotubes. Biochim. Biophys. 2017, 1864, 806–813. [Google Scholar] [CrossRef]
- Vandebrouck, C.; Martin, D.; van Schoor, M.C.; Debaix, H.; Gailly, P. Involvement of TRPC in the Abnormal Calcium Influx Observed in Dystrophic (Mdx) Mouse Skeletal Muscle Fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin Is a Growth-Hormone-Releasing Acylated Peptide from Stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Morris, A. Pancreatic GLP1 Is Involved in Glucose Regulation. Nat. Rev. Endocrinol. 2017, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Katsurada, K.; Yada, T. Neural Effects of Gut- and Brain-Derived Glucagon-like Peptide-1 and Its Receptor Agonist. J. Diabetes Investig. 2016, 7, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Smeets, A.J.; Westerterp-Plantenga, M.S. The Acute Effects of a Lunch Containing Capsaicin on Energy and Substrate Utilisation, Hormones, and Satiety. Eur. J. Nutr. 2009, 48, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Westerterp-Plantenga, M.S.; Smeets, A.; Lejeune, M.P.G. Sensory and Gastrointestinal Satiety Effects of Capsaicin on Food Intake. Int. J. Obes. 2004, 29, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Janssens, P.L.H.R.; Hursel, R.; Westerterp-Plantenga, M.S. Capsaicin Increases Sensation of Fullness in Energy Balance, and Decreases Desire to Eat after Dinner in Negative Energy Balance. Appetite 2014, 77, 44–49. [Google Scholar] [CrossRef]
- Ludy, M.J.; Mattes, R.D. The Effects of Hedonically Acceptable Red Pepper Doses on Thermogenesis and Appetite. Physiol. Behav. 2011, 102, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Camacho, S.; Michlig, S.; de Senarclens-Bezençon, C.; Meylan, J.; Meystre, J.; Pezzoli, M.; Markram, H.; le Coutre, J. Anti-Obesity and Anti-Hyperglycemic Effects of Cinnamaldehyde via Altered Ghrelin Secretion and Functional Impact on Food Intake and Gastric Emptying. Sci. Rep. 2015, 5, 7919. [Google Scholar] [CrossRef] [Green Version]
- Ohara, K.; Fukuda, T.; Ishida, Y.; Takahashi, C.; Ohya, R.; Katayama, M.; Uchida, K.; Tominaga, M.; Nagai, K. β-Eudesmol, an Oxygenized Sesquiterpene, Stimulates Appetite via TRPA1 and the Autonomic Nervous System. Sci. Rep. 2017, 7, 15785. [Google Scholar] [CrossRef]
- Wang, P.; Yan, Z.; Zhong, J.; Chen, J.; Ni, Y.; Li, L.; Ma, L.; Zhao, Z.; Liu, D.; Zhu, Z. Transient Receptor Potential Vanilloid 1 Activation Enhances Gut Glucagon-like Peptide-1 Secretion and Improves Glucose Homeostasis. Diabetes 2012, 61, 2155–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The Arcuate Nucleus Mediates GLP-1 Receptor Agonist Liraglutide-Dependent Weight Loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, F.; Gunawan, A.L.; Tso, P.; Aponte, G.W. Glucagon-like Peptide 1 and Glucose-Dependent Insulinotropic Polypeptide Stimulate Release of Substance P from TRPV1- and TRPA1-Expressing Sensory Nerves. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G23–G35. [Google Scholar] [CrossRef]
- Emery, E.C.; Diakogiannaki, E.; Gentry, C.; Psichas, A.; Habib, A.M.; Bevan, S.; Fischer, M.J.M.; Reimann, F.; Gribble, F.M. Stimulation of GLP-1 Secretion Downstream of the Ligand-Gated Ion Channel TRPA1. Diabetes 2015, 64, 1202–1210. [Google Scholar] [CrossRef] [Green Version]
- Chepurny, O.G.; Holz, G.G.; Roe, M.W.; Leech, C.A. GPR119 Agonist AS1269574 Activates TRPA1 Cation Channels to Stimulate GLP-1 Secretion. Mol. Endocrinol. 2016, 30, 614–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, P.; Mahajan, N.; Singh, D.P.; Kumar, V.; Kumar, V.; Mangal, P.; Boparai, R.K.; Gesing, A.; Bhadada, S.K.; Sharma, S.S.; et al. Allicin, a Dietary Trpa1 Agonist, Prevents High Fat Diet-Induced Dysregulation of Gut Hormones and Associated Complications. Food Funct. 2021, 12, 11526–11536. [Google Scholar] [CrossRef]
- Korner, J.; Chua, S.C.; Williams, J.A.; Leibel, R.L.; Wardlaw, S.L. Regulation of Hypothalamic Proopiomelanocortin by Leptin in Lean and Obese Rats. Neuroendocrinology 1999, 70, 377–383. [Google Scholar] [CrossRef]
- Lee, S.J.; Verma, S.; Simonds, S.E.; Kirigiti, M.A.; Kievit, P.; Lindsley, S.R.; Loche, A.; Smith, M.S.; Cowley, M.A.; Grove, K.L. Leptin Stimulates Neuropeptide Y and Cocaine Amphetamine-Regulated Transcript Coexpressing Neuronal Activity in the Dorsomedial Hypothalamus in Diet-Induced Obese Mice. J. Neurosci. 2013, 33, 15306–15317. [Google Scholar] [CrossRef] [Green Version]
- Mercer, A.J.; Stuart, R.C.; Attard, C.A.; Otero-Corchon, V.; Nillni, E.A.; Low, M.J. Temporal Changes in Nutritional State Affect Hypothalamic POMC Peptide Levels Independently of Leptin in Adult Male Mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, 904–915. [Google Scholar] [CrossRef] [Green Version]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Löllmann, B.; Lowell, B.B.; Flier, J.S. Leptin Levels Reflect Body Lipid Content in Mice: Evidence for Diet-Induced Resistance to Leptin Action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin Levels in Human and Rodent: Measurement of Plasma Leptin and Ob RNA in Obese and Weight-Reduced Subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pei, J.H.; Kuang, J.; Chen, H.M.; Chen, Z.; Li, Z.W.; Yang, H.Z. Effect of Lifestyle Intervention in Patients with Type 2 Diabetes: A Meta-Analysis. Metabolism 2015, 64, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Zsombok, A.; Jiang, Y.; Gao, H.; Anwar, I.J.; Rezai-Zadeh, K.; Enix, C.L.; Münzberg, H.; Derbenev, A.V. Regulation of Leptin Receptor-Expressing Neurons in the Brainstem by TRPV1. Physiol. Rep. 2014, 2, e12160. [Google Scholar] [CrossRef]
- Zsombok, A.; Bhaskaran, M.D.; Gao, H.; Derbenev, A.V.; Smith, B.N. Functional Plasticity of Central TRPV1 Receptors in Brainstem Dorsal Vagal Complex Circuits of Streptozotocin-Treated Hyperglycemic Mice. J. Neurosci. 2011, 31, 14024–14031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin and Leptin Excite Anorexigenic Pro-Opiomelanocortin Neurones via Activation of TRPC5 Channels. J. Neuroendocrinol. 2018, 30, e12501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, M.; Wayman, G.A.; Zhu, M.; Lambert, T.J.; Davare, M.A.; Appleyard, S.M. Leptin-Induced Spine Formation Requires TrpC Channels and the CaM Kinase Cascade in the Hippocampus. J. Neurosci. 2014, 34, 10022–10033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Avshalumov, M.V.; Patel, J.C.; Lee, C.R.; Miller, E.W.; Chang, C.J.; Rice, M.E. Mitochondria Are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 2009, 29, 9002–9010. [Google Scholar] [CrossRef]
- Infanger, D.W.; Sharma, R.V.; Davisson, R.L. NADPH Oxidases of the Brain: Distribution, Regulation, and Function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar] [CrossRef]
- Leloup, C.; Magnan, C.; Benani, A.; Bonnet, E.; Alquier, T.; Offer, G.; Carriere, A.; Périquet, A.; Fernandez, Y.; Ktorza, A.; et al. Mitochondrial Reactive Oxygen Species Are Required for Hypothalamic Glucose Sensing. Diabetes 2006, 55, 2084–2090. [Google Scholar] [CrossRef] [Green Version]
- Benani, A.; Troy, S.; Carmona, M.C.; Fioramonti, X.; Lorsignol, A.; Leloup, C.; Casteilla, L.; Pénicaud, L. Role for Mitochondrial Reactive Oxygen Species in Brain Lipid Sensing: Redox Regulation of Food Intake. Diabetes 2007, 56, 152–160. [Google Scholar] [CrossRef]
- Diano, S.; Liu, Z.W.; Jeong, J.K.; Dietrich, M.O.; Ruan, H.B.; Kim, E.; Suyama, S.; Kelly, K.; Gyengesi, E.; Arbiser, J.L.; et al. Peroxisome Proliferation–Associated Control of Reactive Oxygen Species Sets Melanocortin Tone and Feeding in Diet-Induced Obesity. Nat. Med. 2011, 17, 1121–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belza, A.; Jessen, A.B. Bioactive Food Stimulants of Sympathetic Activity: Effect on 24-h Energy Expenditure and Fat Oxidation. Eur. J. Clin. Nutr. 2005, 59, 733–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belza, A.; Frandsen, E.; Kondrup, J. Body Fat Loss Achieved by Stimulation of Thermogenesis by a Combination of Bioactive Food Ingredients: A Placebo-Controlled, Double-Blind 8-Week Intervention in Obese Subjects. Int. J. Obes. 2006, 31, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Reinbach, H.C.; Smeets, A.; Martinussen, T.; Møller, P.; Westerterp-Plantenga, M.S. Effects of Capsaicin, Green Tea and CH-19 Sweet Pepper on Appetite and Energy Intake in Humans in Negative and Positive Energy Balance. Clin. Nutr. 2009, 28, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Belza, A.; Gille, M.B.; Schultz John, S.; Kondrup, J. The Beta-Adrenergic Antagonist Propranolol Partly Abolishes Thermogenic Response to Bioactive Food Ingredients. Metab. Clin. Exp. 2009, 58, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Janssens, P.L.H.R.; Hursel, R.; Martens, E.A.P.; Westerterp-Plantenga, M.S. Acute Effects of Capsaicin on Energy Expenditure and Fat Oxidation in Negative Energy Balance. PLoS ONE 2013, 8, e67786. [Google Scholar] [CrossRef] [Green Version]
- Rigamonti, A.E.; Casnici, C.; Marelli, O.; de Col, A.; Tamini, S.; Lucchetti, E.; Tringali, G.; de Micheli, R.; Abbruzzese, L.; Bortolotti, M.; et al. Acute Administration of Capsaicin Increases Resting Energy Expenditure in Young Obese Subjects without Affecting Energy Intake, Appetite, and Circulating Levels of Orexigenic/Anorexigenic Peptides. Nutr. Res. 2018, 52, 71–79. [Google Scholar] [CrossRef]
- Chaiyasit, K.; Khovidhunkit, W.; Wittayalertpanya, S. Pharmacokinetic and The Effect of Capsaicin in Capsicum Frutescens on Decreasing Plasma Glucose Level. J. Med. Assoc. Thail. 2009, 92, 108–113. [Google Scholar]
- Kobata, K.; Todo, T.; Yazawa, S.; Iwai, K.; Watanabe, T. Novel Capsaicinoid-like Substances, Capsiate and Dihydrocapsiate, from the Fruits of a Nonpungent Cultivar, CH-19 Sweet, of Pepper (Capsicum annuum L.). J. Agric. Food Chem. 1998, 46, 1695–1697. [Google Scholar] [CrossRef]
- Snitker, S.; Fujishima, Y.; Shen, H.; Ott, S.; Pi-Sunyer, X.; Furuhata, Y.; Sato, H.; Takahashi, M. Effects of Novel Capsinoid Treatment on Fatness and Energy Metabolism in Humans: Possible Pharmacogenetic Implications. Am. J. Clin. Nutr. 2009, 89, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Galgani, J.E.; Ravussin, E. Effect of Dihydrocapsiate on Resting Metabolic Rate in Humans. Am. J. Clin. Nutr. 2010, 92, 1089–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomer, R.J.; Canale, R.E.; Shastri, S.; Suvarnapathki, S. Effect of Oral Intake of Capsaicinoid Beadlets on Catecholamine Secretion and Blood Markers of Lipolysis in Healthy Adults: A Randomized, Placebo Controlled, Double-Blind, Cross-over Study. Lipids Health Dis. 2010, 9, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneshiro, T.; Aita, S.; Kawai, Y.; Iwanaga, T.; Saito, M. Nonpungent Capsaicin Analogs (Capsinoids) Increase Energy Expenditure through the Activation of Brown Adipose Tissue in Humans. Am. J. Clin. Nutr. 2012, 95, 845–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirengi, S.; Homma, T.; Inoue, N.; Sato, H.; Yoneshiro, T.; Matsushita, M.; Kameya, T.; Sugie, H.; Tsuzaki, K.; Saito, M.; et al. Assessment of Human Brown Adipose Tissue Density during Daily Ingestion of Thermogenic Capsinoids Using Near-Infrared Time-Resolved Spectroscopy. J. Biomed. Opt. 2016, 21, 913051–913057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, P.V.; Gan, S.H. Cinnamon: A Multifaceted Medicinal Plant. Evid. Based Complement. Altern. Med. 2014, 2014, 642942. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Safdar, M.; Ali Khan, M.M.; Khattak, K.N.; Anderson, R.A. Cinnamon Improves Glucose and Lipids of People with Type 2 Diabetes. Diabetes Care 2003, 26, 3215–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussel, A.M.; Hininger, I.; Benaraba, R.; Ziegenfuss, T.N.; Anderson, R.A. Antioxidant Effects of a Cinnamon Extract in People with Impaired Fasting Glucose That Are Overweight or Obese. J. Am. Coll. Nutr. 2013, 28, 16–21. [Google Scholar] [CrossRef]
- Liu, Y.; Cotillard, A.; Vatier, C.; Bastard, J.P.; Fellahi, S.; Stévant, M.; Allatif, O.; Langlois, C.; Bieuvelet, S.; Brochot, A.; et al. A Dietary Supplement Containing Cinnamon, Chromium and Carnosine Decreases Fasting Plasma Glucose and Increases Lean Mass in Overweight or Obese Pre-Diabetic Subjects: A Randomized, Placebo-Controlled Trial. PLoS ONE 2015, 10, e0138646. [Google Scholar] [CrossRef] [PubMed]
- Gupta Jain, S.; Puri, S.; Misra, A.; Gulati, S.; Mani, K. Effect of Oral Cinnamon Intervention on Metabolic Profile and Body Composition of Asian Indians with Metabolic Syndrome: A Randomized Double -Blind Control Trial. Lipids Health Dis. 2017, 16, 113. [Google Scholar] [CrossRef]
- Zare, R.; Nadjarzadeh, A.; Zarshenas, M.M.; Shams, M.; Heydari, M. Efficacy of Cinnamon in Patients with Type II Diabetes Mellitus: A Randomized Controlled Clinical Trial. Clin. Nutr. 2019, 38, 549–556. [Google Scholar] [CrossRef]
- Lu, T.; Sheng, H.; Wu, J.; Cheng, Y.; Zhu, J.; Chen, Y. Cinnamon Extract Improves Fasting Blood Glucose and Glycosylated Hemoglobin Level in Chinese Patients with Type 2 Diabetes. Nutr. Res. 2012, 32, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Beejmohun, V.; Peytavy-Izard, M.; Mignon, C.; Muscente-Paque, D.; Deplanque, X.; Ripoll, C.; Chapal, N. Acute Effect of Ceylon Cinnamon Extract on Postprandial Glycemia: Alpha-Amylase Inhibition, Starch Tolerance Test in Rats, and Randomized Crossover Clinical Trial in Healthy Volunteers. BMC Complement. Altern. Med. 2014, 14, 351. [Google Scholar] [CrossRef] [Green Version]
- Suppapitiporn, S.; Kanpaksi, N.; Suppapitiporn, S. The Effect of Cinnamon Cassia Powder in Type 2 Diabetes Mellitus. J. Med. Assoc. Thail. 2006, 89 (Suppl. S3), S200–S205. [Google Scholar]
- Talaei, B.; Amouzegar, A.; Sahranavard, S.; Hedayati, M.; Mirmiran, P.; Azizi, F. Effects of Cinnamon Consumption on Glycemic Indicators, Advanced Glycation End Products, and Antioxidant Status in Type 2 Diabetic Patients. Nutrients 2017, 9, 991. [Google Scholar] [CrossRef] [Green Version]
- Vanschoonbeek, K.; Thomassen, B.J.W.; Senden, J.M.; Wodzig, W.K.W.H.; van Loon, L.J.C. Cinnamon Supplementation Does Not Improve Glycemic Control in Postmenopausal Type 2 Diabetes Patients. J. Nutr. 2006, 136, 977–980. [Google Scholar] [CrossRef] [Green Version]
- Markey, O.; McClean, C.M.; Medlow, P.; Davison, G.W.; Trinick, T.R.; Duly, E.; Shafat, A. Effect of Cinnamon on Gastric Emptying, Arterial Stiffness, Postprandial Lipemia, Glycemia, and Appetite Responses to High-Fat Breakfast. Cardiovasc. Diabetol. 2011, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Akilen, R.; Tsiami, A.; Devendra, D.; Robinson, N. Glycated Haemoglobin and Blood Pressure-Lowering Effect of Cinnamon in Multi-Ethnic Type 2 Diabetic Patients in the UK: A Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Diabet. Med. 2010, 27, 1159–1167. [Google Scholar] [CrossRef]
- Uchida, K.; Tominaga, M. The Role of Thermosensitive TRP (Transient Receptor Potential) Channels in Insulin Secretion. Endocr. J. 2011, 58, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Colsoul, B.; Nilius, B.; Vennekens, R. Transient Receptor Potential (TRP) Cation Channels in Diabetes. Curr. Top. Med. Chem. 2013, 13, 258–269. [Google Scholar] [CrossRef]
- Ali, E.S.; Rychkov, G.Y.; Barritt, G.J. Trpm2 Non-Selective Cation Channels in Liver Injury Mediated by Reactive Oxygen Species. Antioxidants 2021, 10, 1243. [Google Scholar] [CrossRef]
- Kheradpezhouh, E.; Barritt, G.J.; Rychkov, G.Y. Curcumin Inhibits Activation of TRPM2 Channels in Rat Hepatocytes. Redox Biol. 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
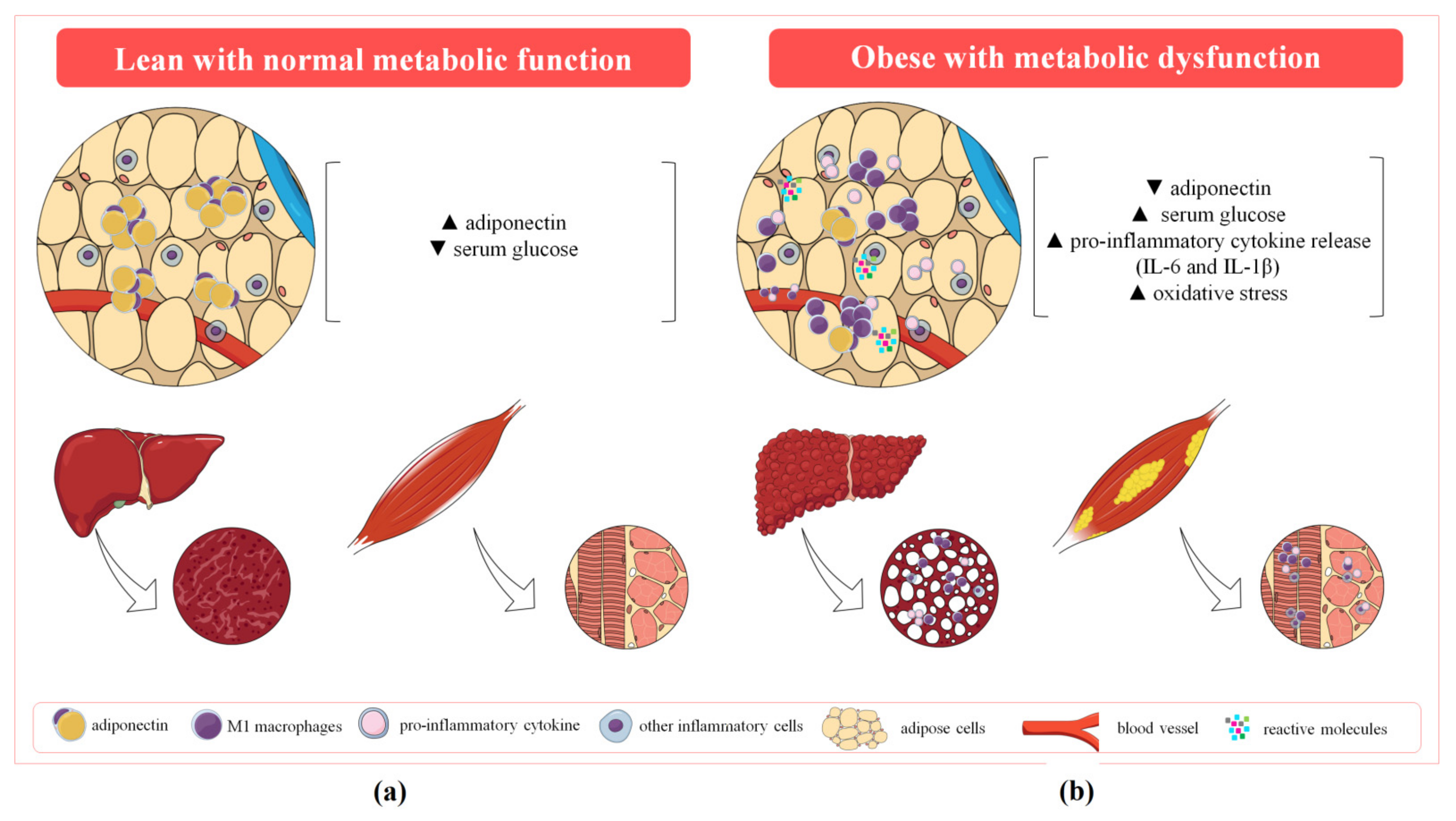{kind=link}
{kind=link}
{kind=link}
| TRP Channel | Reactive Molecule | Cell Type | Activation Mode | Ca2+ Influx | Electrophysiology |
|---|---|---|---|---|---|
| TRPV1 | H2O2 | HEK293T [123,150,152] | Sensitization | ✓ | ✓ |
| Bovine aortic endothelial cells [123] | Sensitization | ✓ | |||
| TRPA1 | H2O2 | HEK293T [157,158,159] | Direct | ✓ | |
| DRG neurones [158,159,160] | Direct | ✓ | |||
| Bladder neuronal afferents [161] | Direct | ✓ | |||
| CHO cells [160] | Direct | ✓ | |||
| NO | HEK293T [159] | Direct | ✓ | ||
| DRG neurones [159] | Direct | ✓ | |||
| H+ | HEK293T [159] | Direct | ✓ | ||
| DRG neurones [159] | Direct | ✓ | |||
| Aldehydes (4-HNE and 4-ONE) | HEK293T cells [157] | Direct | ✓ | ||
| DRG and trigeminal ganglia neurones [157,160] | Direct | ✓ | |||
| CHO cells [160,162] | Direct | ✓ | |||
| TRPC5 | H2O2 | HEK293T cells [145] | Direct | ✓ | |
| Reduced TRX | HEK293T cells [150] | Direct | ✓ | ||
| Synoviocytes [150] | Direct | ✓ |
| TRP Channel | Cell/Tissue | PCR/ qPCR | Immunostaining/ Immunofluorescence | Western Blot | Ca2+ Influx | Electrophysiology |
|---|---|---|---|---|---|---|
| TRPV1 | adipose tissue/adipocytes [124,125,200,201] | ✓ | ✓ | ✓ | ✓ | |
| liver [126,127] | ✓ | ✓ | ✓ | ✓ | ||
| M1 macrophages [128] | ✓ | ✓ | ||||
| pancreatic β-cells/langerhans islets [132] | ✓ | ✓ | ✓ | |||
| coronary endothelial cells [123] | ✓ | |||||
| T cells [129,130,131] | ✓ | ✓ | ✓ | ✓ | ||
| skeletal muscle [202,203,204] | ✓ | ✓ | ✓ | ✓ | ||
| pro-opiomelanocortin neurones [205] | ✓ | ✓ | ✓ | ✓ | ||
| TRPA1 | pancreatic β-cells/langerhans islets [137,206] | ✓ | ✓ | ✓ | ✓ | ✓ |
| T cells [175,176,177] | ✓ | ✓ | ✓ | ✓ | ✓ | |
| skeletal muscle cells [207] | ✓ | ✓ | ✓ | ✓ | ✓ | |
| monocytes/macrophages [179,180] | ✓ | ✓ | ✓ | ✓ | ||
| TRPC5 | endothelial cells [166] | ✓ | ||||
| T cells [185] | ✓ | ✓ | ||||
| M1 macrophages [186] | ✓ | ✓ | ||||
| pancreas [208] | ✓ | |||||
| adipose tissue [147,200] | ✓ | ✓ | ✓ | ✓ | ✓ | |
| pro-opiomelanocortin neurones [209,210,211] | ✓ |
| TRP Channel | Endogenous Agonists | Expression Site | Role in MS |
|---|---|---|---|
| TRPV1 | 12 (S)-HPETE [118], 20-HETE [119], 9-HODE and 13-HODE [120], anandamide [121], H2S [122], ROS (H2O2) [123] | Adipose tissue/adipocytes [124,125,200,201], liver [126,127], M1 macrophages [128], pancreatic β-cells/langerhans islets [132], coronary endothelial cells [123], T cells [129,130,131], skeletal muscle [202,203,204], pro-opiomelanocortin neurones [205] | Increase of insulin sensitivity [216,220,221], browning of WAT, reduction of lipid synthesis and obesity/adiposity [201,203,204,239], enhanced thermogenesis [240] and leptin sensitivity [219], reduction of lipid accumulation and TG [127], protection against endothelial dysfunction [253], increase of GLP-1 and attenuation of ghrelin production [278] |
| TRPA1 | Methylglyoxal [138], 4-HNE, 15-deoxy-delta(12,14)-prosta-glandin J2 (15d-PGJ2) and H2O2 [139] | Pancreatic β-cells/langerhans islets [137,206], T cells [175,176,177], adipocytes [244,245], vagus nerve [246] | Macrophage-mediate responses in atherosclerosis [180,184], increase of insulin secretion [137,222,223,224,225] and sensitivity [228,258,259], reduction of insulin signalling and insulin-induced glucose uptake in skeletal muscle cells [229], weight loss and reduction of TG and cholesterol [244,247], attenuated adipogenesis [250], increased adipose tissue inflammation and ROS [248,250] reduction of ghrelin [282], production of ghrelin [288] |
| TRPC5 | H2O2 [145], reduced TRX [146], and fatty acids [147] | Endothelial cells [166], T cells [185], M1 macrophages [186], pancreas [208], adipose tissue [147,200], pro-opiomelanocortin neurones [209,210,211] | Polarization of macrophages to M2 and protection against atherosclerosis [186], negative regulation of adiponectin [147], enhance of energy expenditure [209,298] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo, M.C.; Soczek, S.H.S.; Pontes, J.P.; Marques, L.A.C.; Santos, G.S.; Simão, G.; Bueno, L.R.; Maria-Ferreira, D.; Muscará, M.N.; Fernandes, E.S. An Overview of the TRP-Oxidative Stress Axis in Metabolic Syndrome: Insights for Novel Therapeutic Approaches. Cells 2022, 11, 1292. https://doi.org/10.3390/cells11081292
Araújo MC, Soczek SHS, Pontes JP, Marques LAC, Santos GS, Simão G, Bueno LR, Maria-Ferreira D, Muscará MN, Fernandes ES. An Overview of the TRP-Oxidative Stress Axis in Metabolic Syndrome: Insights for Novel Therapeutic Approaches. Cells. 2022; 11(8):1292. https://doi.org/10.3390/cells11081292
Chicago/Turabian StyleAraújo, Mizael C., Suzany H. S. Soczek, Jaqueline P. Pontes, Leonardo A. C. Marques, Gabriela S. Santos, Gisele Simão, Laryssa R. Bueno, Daniele Maria-Ferreira, Marcelo N. Muscará, and Elizabeth S. Fernandes. 2022. "An Overview of the TRP-Oxidative Stress Axis in Metabolic Syndrome: Insights for Novel Therapeutic Approaches" Cells 11, no. 8: 1292. https://doi.org/10.3390/cells11081292
APA StyleAraújo, M. C., Soczek, S. H. S., Pontes, J. P., Marques, L. A. C., Santos, G. S., Simão, G., Bueno, L. R., Maria-Ferreira, D., Muscará, M. N., & Fernandes, E. S. (2022). An Overview of the TRP-Oxidative Stress Axis in Metabolic Syndrome: Insights for Novel Therapeutic Approaches. Cells, 11(8), 1292. https://doi.org/10.3390/cells11081292