
Autophagy in Rheumatic Diseases: Role in the Pathogenesis and Therapeutic Approaches

 and
and
Abstract
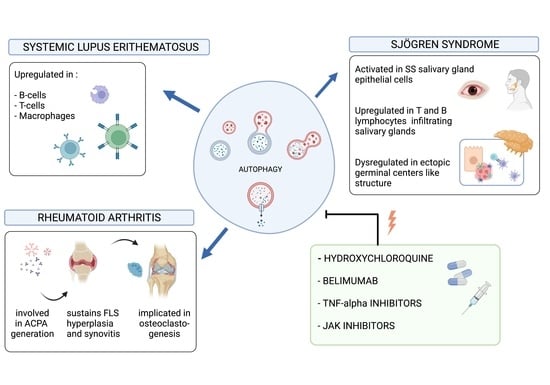
:
{kind=link}
{kind=link}
1. Introduction
2. Rheumatoid Arthritis
3. Systemic Lupus Erythematosus
4. Sjögren’s Syndrome
5. Autophagy Pathway Modulating Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 2011, 240, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munz, C. Antigen processing via autophagy-not only for MHC class II presentation anymore? Curr. Opin. Immunol. 2010, 22, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.M.; Edinger, A.L. The complex interplay between autophagy, apoptosis, and necrotic signals promotes T-cell homeostasis. Immunol. Rev. 2010, 236, 95–109. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Catrina, A.; Krishnamurthy, A.; Rethi, B. Current view on the pathogenic role of anti-citrullinated protein antibodies in rheumatoid arthritis. RMD Open 2021, 7, e001228. [Google Scholar] [CrossRef]
- Sorice, M.; Iannuccelli, C.; Manganelli, V.; Capozzi, A.; Alessandri, C.; Lococo, E.; Garofalo, T.; Di Franco, M.; Bombardieri, M.; Nerviani, A. Autophagy generates citrullinated peptides in human synoviocytes: A possible trigger for anti-citrullinated peptide antibodies. Rheumatology 2016, 55, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jiang, P.; Guo, S.; Schrodi, S.J.; He, D. Apoptosis, autophagy, NETosis, Necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front. Immunol. 2021, 12, 809806. [Google Scholar] [CrossRef]
- Liu, H.; Pope, R.M. The role of apoptosis in rheumatoid arthritis. Curr. Opin. Pharmacol. 2003, 3, 317–322. [Google Scholar] [CrossRef]
- Pap, T.; Müller-Ladner, U.; Gay, R.E.; Gay, S. Fibroblast biology. Role of synovialfibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000, 2, 361–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baier, A.; Meineckel, I.; Gay, S.; Pap, T. Apoptosis in rheumatoid arthritis. Curr. Opin. Rheumatol. 2003, 15, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Xu, P.; Yao, J.-F.; Zhang, Y.-G.; Hou, W.-K.; Lu, S.-M. Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis. Inflamm. Res. 2013, 62, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Ospelt, C.; Gay, R.E.; Gay, S.; Klein, K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2014, 66, 40–48. [Google Scholar] [CrossRef]
- Shin, Y.J.; Han, S.-H.; Kim, D.-S.; Lee, G.-H.; Yoo, W.-H.; Kang, Y.-M.; Choi, J.-Y.; Lee, Y.-C.; Park, S.J.; Jeong, S.-K.; et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res. Ther. 2010, 12, R19. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Wang, H.; Wu, Y.; He, Z.; Qin, Y.; Shen, Q. The autophagy level is increased in the synovial tissues of patients with active rheumatoid arthritis and is correlated with disease severity. Mediat. Inflamm. 2017, 2017, 7623145. [Google Scholar] [CrossRef]
- Lin, N.Y.; Beyer, C.; Giessl, A.; Kireva, T.; Scholtysek, C.; Uderhardt, S.; Munoz, L.E.; Dees, C.; Distler, A.; Wirtz, S.; et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 2013, 72, 761–768. [Google Scholar] [CrossRef]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. Autophagy in autoimmune disease. J. Mol. Med. 2015, 93, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Ciccacci, C.; Perricone, C.; Alessandri, C.; Latini, A.; Politi, C.; Delunardo, F.; Pierdominici, M.; Conti, F.; Novelli, G.; Ortona, E.; et al. Evaluation of ATG5 polymorphisms in Italian patients with systemic lupus erythematosus: Contribution to disease susceptibility and clinical phenotypes. Lupus 2018, 27, 1464–1469. [Google Scholar] [CrossRef]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.N.; Gilpin, C.; Levine, B. Autophagy gene- dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, N.J.; Lees, M.J.; Gabriel, L.; Maniati, E.; Rose, S.J.; Potter, P.K.; Morley, B.J. A defect in Marco expression contributes to systemic lupus erythematosus development via failure to clear apoptotic cells. J. Immunol. 2009, 182, 1982–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, F.; Spinelli, F.R.; Alessandri, C.; Valesini, G. Toll-like receptors and lupus nephritis. Clin. Rev. Allergy Immunol. 2011, 40, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Jacquel, A.; Obba, S.; Boyer, L.; Dufies, M.; Robert, G.; Gounon, P.; Lemichez, E.; Luciano, F.; Solary, E.; Auberger, P. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocyt- ic functions. Blood 2012, 119, 4527–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Yue, Y.; Dong, C.; Shi, Y.; Xiong, S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin. Exp. Rheumatol. 2014, 32, 705–714. [Google Scholar]
- Möckel, T.; Basta, F.; Weinmann-Menke, J.; Schwarting, A. B cell activating factor (BAFF): Structure, functions, autoimmunity and clinical implications in Systemic Lupus Erythematosus (SLE). Autoimmun. Rev. 2021, 20, 102736. [Google Scholar] [CrossRef]
- Pers, J.O.; Daridon, C.; Devauchelle, V.; Jousse, S.; Sarauz, A.; Jamin, C.; Youinou, P. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1050, 34–39. [Google Scholar] [CrossRef]
- Petri, M.A.; van Vollenhoven, R.F.; Buyon, J.; Levy, R.A.; Navarra, S.V.; Cervera, R.; Zhong, Z.J.; Freimuth, W.W.; Bliss Group. Baseline predictors of systemic lupus erythematosus flares: Data from the combined placebo groups in the phase III belimumab trials. Arthritis Rheum. 2013, 65, 2143–2153. [Google Scholar] [CrossRef]
- Otipoby, K.L.; Sasaki, Y.; Schmidt-Supprian, M.; Patke, A.; Gareus, R.; Pasparakis, M.; Tarakhovsky, A.; Rajewsky, K. BAFF activates Akt and Erk through BAFF-R in an IKK1-dependent manner in primary mouse B cells. Proc. Natl. Acad. Sci. USA 2008, 105, 12435–12438. [Google Scholar] [CrossRef] [Green Version]
- Rickert, R.C.; Jellusova, J.; Miletic, A.V. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol. Rev. 2011, 244, 115–133. [Google Scholar] [CrossRef]
- Alessandri, C.; Barbati, C.; Vacirca, D.; Piscopo, P.; Confaloni, A.; Sanchez, M.; Maselli, A.; Colasanti, T.; Conti, F.; Truglia, S.; et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J. 2012, 26, 4722–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandri, C.; Ciccia, F.; Priori, R.; Astorri, E.; Guggino, G.; Alessandro, R.; Rizzo, A.; Conti, F.; Minniti, A.; Barbati, C.; et al. CD4 T lymphocyte autophagy is upregulated in the salivary glands of primary Sjögren’s syndrome patients and correlates with focus score and disease activity. Arthritis Res. Ther. 2017, 19, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colafrancesco, S.; Vomero, M.; Iannizzotto, V.; Minniti, A.; Barbati, C.; Arienzo, F.; Mastromanno, L.; Colasanti, T.; Izzo, R.; Nayar, S.; et al. Autophagy occurs in lymphocytes infiltrating Sjögren’s syndrome minor salivary glands and correlates with histological severity of salivary gland lesions. Arthritis Res. Ther. 2020, 22, 238. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, H.M. Sjogren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Ji, Y.W.; Shim, J.; Noh, H.; Yeo, A.; Park, C.; Park, M.S.; Chang, E.J.; Lee, H.K. Activation of HIF-1a (hypoxia inducible factor-1a) prevents dry eye-induced acinar cell death in the lacrimal gland. Cell Death Dis. 2014, 5, e1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, Y.S.; Lee, H.J.; Shin, S.; Chung, S.-H. Elevation of autophagy markers in Sjogren syndrome dry eye. Sci. Rep. 2017, 7, 17280. [Google Scholar] [CrossRef] [Green Version]
- Byun, Y.S.; Lee, H.J.; Shin, S.; Choi, M.Y.; Kim, H.S.; Chung, S.H. Tear ATG5 as a potential novel biomarker in the diagnosis of Sjogren syndrome. Diagnostics 2021, 11, 71. [Google Scholar] [CrossRef]
- Morgan-Bathke, M.; Lin, H.H.; Chibly, A.M.; Zhang, W.; Sun, X.; Chen, C.H.; Flodby, P.; Borok, Z.; Wu, R.; Arnett, D.; et al. Deletion of ATG5 shows a role of autophagy in salivary homeostatic control. J. Dent. Res. 2013, 92, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Morgan-Bathke, M.; Hill, G.A.; Harris, Z.I.; Lin, H.H.; Chibly, A.M.; Klein, R.R.; Burd, R.; Ann, D.K.; Limesand, K.H. Autophagy correlates with maintenance of salivary gland function following radiation. Sci. Rep. 2014, 4, 5206. [Google Scholar] [CrossRef] [Green Version]
- Colafrancesco, S.; Barbati, C.; Priori, R.; Putro, E.; Giardina, F.; Gattamelata, A.; Monosi, B.; Colasanti, T.; Celia, A.I.; Cerbelli, B.; et al. Maladaptive autophagy in the pathogenesis of autoimmune epithelitis in Sjögren’s Syndrome. Arthritis Rheumatol. 2021, 74, 654–664. [Google Scholar] [CrossRef]
- Katsiougiannis, S.; Tenta, R.; Skopouli, F.N. Endoplasmic reticulum stress causes autophagy and apoptosis leading to cellular redistribution of the autoantigens Ro/Sjögren’s syndrome-related antigen A (SSA) and La/SSB in salivary gland epithelial cells. Clin. Exp. Immunol. 2015, 181, 244–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wang, F.; Schall, N.; Muller, S. Rescue of autophagy and lysosome defects in salivary glands of MRL/lpr mice by a therapeutic phosphopeptide. J. Autoimmun. 2018, 90, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Voynova, E.; Lefebvre, F.; Qadri, A.; Muller, S. Correction of autophagy impairment inhibits pathology in the NOD.H-2h4 mouse model of primary Sjögren’s syndrome. J. Autoimmun. 2020, 108, 102418. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Edman, M.C.; Janga, S.R.; Yarber, F.; Meng, Z.; Linngam, W.; Bushman, J.; Ma, T.; Liu, S.; Stan, L.; et al. Rapamycin eye drops suppress lacrimal gland inflammation in a murine model of Sjogren’s syndrome. Investig. Ophthalmol. Vis. Sci. 2017, 58, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Harr, M.W.; McColl, K.S.; Zhong, F.; Molitoris, J.K.; Distelhorst, C.W. Glucocorticoids downregulate Fyn and inhibit IP (3)-mediated calcium signaling to promote autophagy in T-lymphocytes. Autophagy 2010, 6, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fan, J.; Lin, Y.S.; Guo, Y.S.; Gao, B.; Shi, Q.Y.; Wei, B.-Y.; Chen, L.; Yang, L.; Liu, J.; et al. Glucocorticoids induce autophagy in rat bone marrow mesenchymal stem cells. Mol. Med. Rep. 2015, 11, 2711–2716. [Google Scholar] [CrossRef]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.W.; Kelly, R.; Winans, T.; Marchena, I.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Tily, H.; Francis, L.; et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial. Lancet 2018, 391, 1186–1196. [Google Scholar] [CrossRef]
- Spinelli, F.R.; Barbati, C.; Cecarelli, F.; Morello, F.; Colasanti, T.; Vomero, M.; Massaro, L.; Orefice, V.; Alessandri, C.; Valesini, G.; et al. B lymphocyte stimulator modulates number and function of endothelial progenitor cells in systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 245. [Google Scholar] [CrossRef] [Green Version]
- Colasanti, T.; Spinelli, F.R.; Barbati, C.; Ceccarelli, F.; Scarpa, S.; Vomero, M.; Alessandri, C.; Valesini, G.; Conti, F. Belimumab decreases autophagy and citrullination in peripheral blood mononuclear cells from patients with systemic Lupus Erythematosus. Cells 2022, 11, 262. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W. Effects of TNF-α on autophagy of rheumatoid arthritis fibroblast-like synoviocytes and regulation of the NF-κB signaling pathway. Immunobiology 2021, 226, 152059. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; Trollmo, C.; Klint, E.; Engstrom, M.; Lampa, J.; Hermansson, Y.; Klareskog, L.; Ulfgren, A.K. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: Extended report. Arthritis Rheum. 2005, 52, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vomero, M.; Manganelli, V.; Barbati, C.; Colasanti, T.; Capozzi, A.; Finucci, A.; Spinelli, F.R.; Ceccarelli, F.; Perricone, C.; Truglia, S.; et al. Reduction of autophagy and increase in apoptosis correlates with a favorable clinical outcome in patients with rheumatoid arthritis treated with anti-TNF drugs. Arthritis Res. Ther. 2019, 21, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billah, M.; Ridiandries, A.; Allahwala, U.K.; Mudaliar, H.; Dona, A.; Hunyor, S.; Khachigian, L.M.; Bhindi, R. Remote ischemic preconditioning induces cardioprotective autophagy and signals through the IL-6-dependent JAK-STAT Pathway. Int. J. Mol. Sci. 2020, 21, 1692. [Google Scholar] [CrossRef] [Green Version]
- Ojha, R.; Singh, S.K.; Bhattacharyya, S. JAK-mediated autophagy regulates stemness and cell survival in cisplatin resistant bladder cancer cells. Biochim. Biophys. Acta 2016, 1860 11 Pt A, 2484–2497. [Google Scholar] [CrossRef]
- Zhang, L.J.; Ni, S.Z.; Zhou, X.L.; Zhao, Y. Hemorrhagic shock sensitized the diaphragm to ventilator-induced dysfunction through the activation of IL-6/JAK/STAT signaling-mediated autophagy in rats. Mediat. Inflamm. 2019, 14, 3738409. [Google Scholar] [CrossRef]
- Li, H.; Bi, Q.; Cui, H.; Lv, C.; Wang, M. Suppression of autophagy through JAK2/STAT3 contributes to the therapeutic action of rhynchophylline on asthma. BMC Complement. Med. Ther. 2021, 21, 21. [Google Scholar] [CrossRef]
- Chen, M.; Li, M.; Zhang, N.; Sun, W.; Wang, H.; Wei, W. Mechanism of miR-218-5p in autophagy, apoptosis and oxidative stress in rheumatoid arthritis synovial fibroblasts is mediated by KLF9 and JAK/STAT3 pathways. J. Investig. Med. 2021, 69, 824–832. [Google Scholar] [CrossRef]
- Aota, K.; Yamanoi, T.; Kani, K.; Ono, S.; Momota, Y.; Azuma, M. Inhibition of JAK-STAT signaling by baricitinib reduces interferon-γ-induced CXCL10 production in human salivary gland ductal cells. Inflammation 2021, 44, 206–216. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celia, A.I.; Colafrancesco, S.; Barbati, C.; Alessandri, C.; Conti, F. Autophagy in Rheumatic Diseases: Role in the Pathogenesis and Therapeutic Approaches. Cells 2022, 11, 1359. https://doi.org/10.3390/cells11081359
Celia AI, Colafrancesco S, Barbati C, Alessandri C, Conti F. Autophagy in Rheumatic Diseases: Role in the Pathogenesis and Therapeutic Approaches. Cells. 2022; 11(8):1359. https://doi.org/10.3390/cells11081359
Chicago/Turabian StyleCelia, Alessandra Ida, Serena Colafrancesco, Cristiana Barbati, Cristiano Alessandri, and Fabrizio Conti. 2022. "Autophagy in Rheumatic Diseases: Role in the Pathogenesis and Therapeutic Approaches" Cells 11, no. 8: 1359. https://doi.org/10.3390/cells11081359
APA StyleCelia, A. I., Colafrancesco, S., Barbati, C., Alessandri, C., & Conti, F. (2022). Autophagy in Rheumatic Diseases: Role in the Pathogenesis and Therapeutic Approaches. Cells, 11(8), 1359. https://doi.org/10.3390/cells11081359