
Fas/CD95 Signaling Pathway in Damage-Associated Molecular Pattern (DAMP)-Sensing Receptors

 ,
,  ,
, 
Abstract
:1. Introduction
2. CD95 and DAMP-Sensor-Mediated Pathologies
2.1. Genetic Mutations in CD95 and DAMP Sensors and Chronic Inflammatory Diseases
2.1.1. CD95-Associated Genetic Disorders
2.1.2. DAMP-Sensor-Associated Genetic Disorders
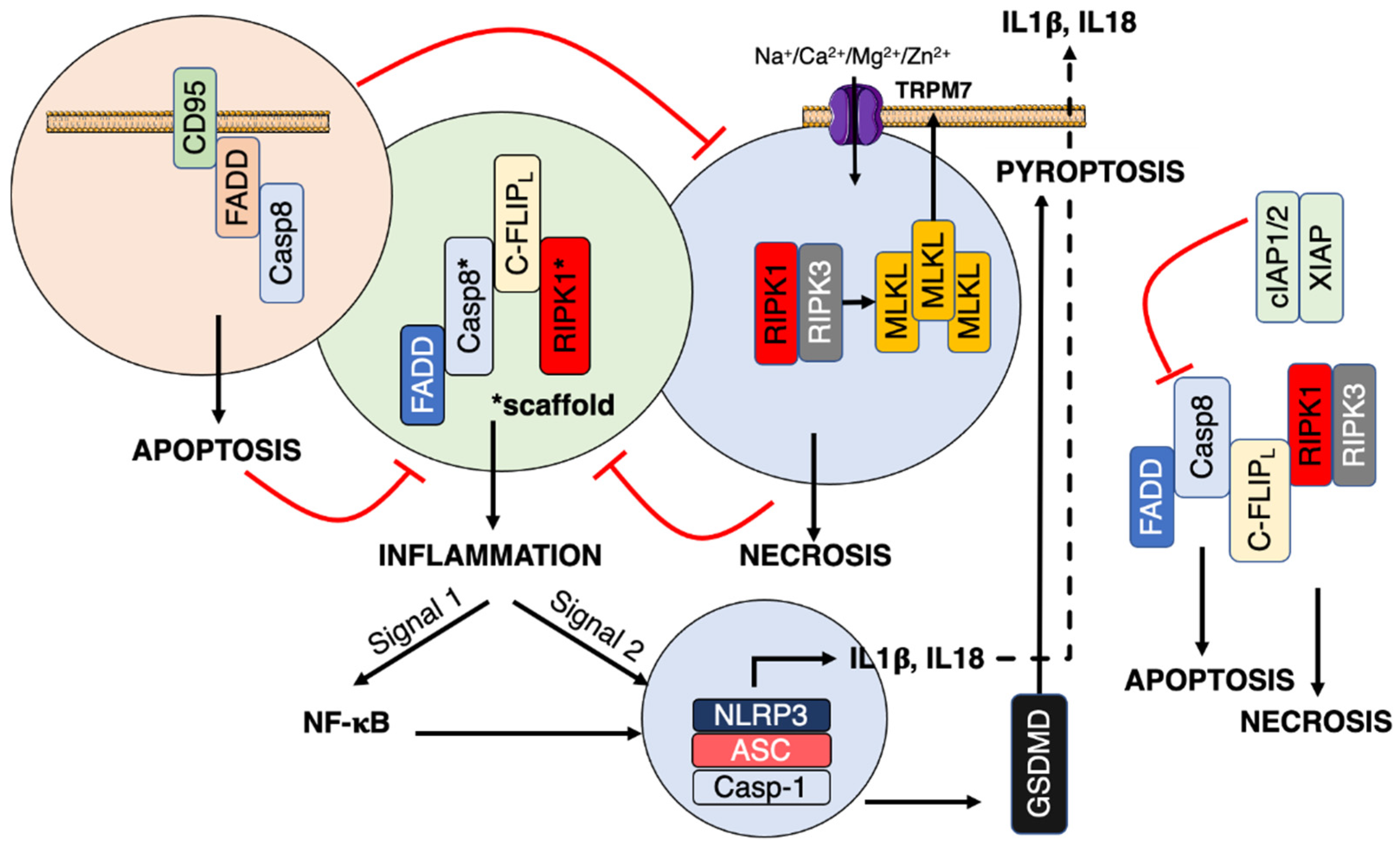
3. CD95/Fas
3.1. Classical Apoptotic Program
3.2. Necroptosis
3.3. Non-Apoptotic Signals
4. Inflammation
4.1. Inflammasome
4.2. PAMP Sensors and CD95 Crosstalk
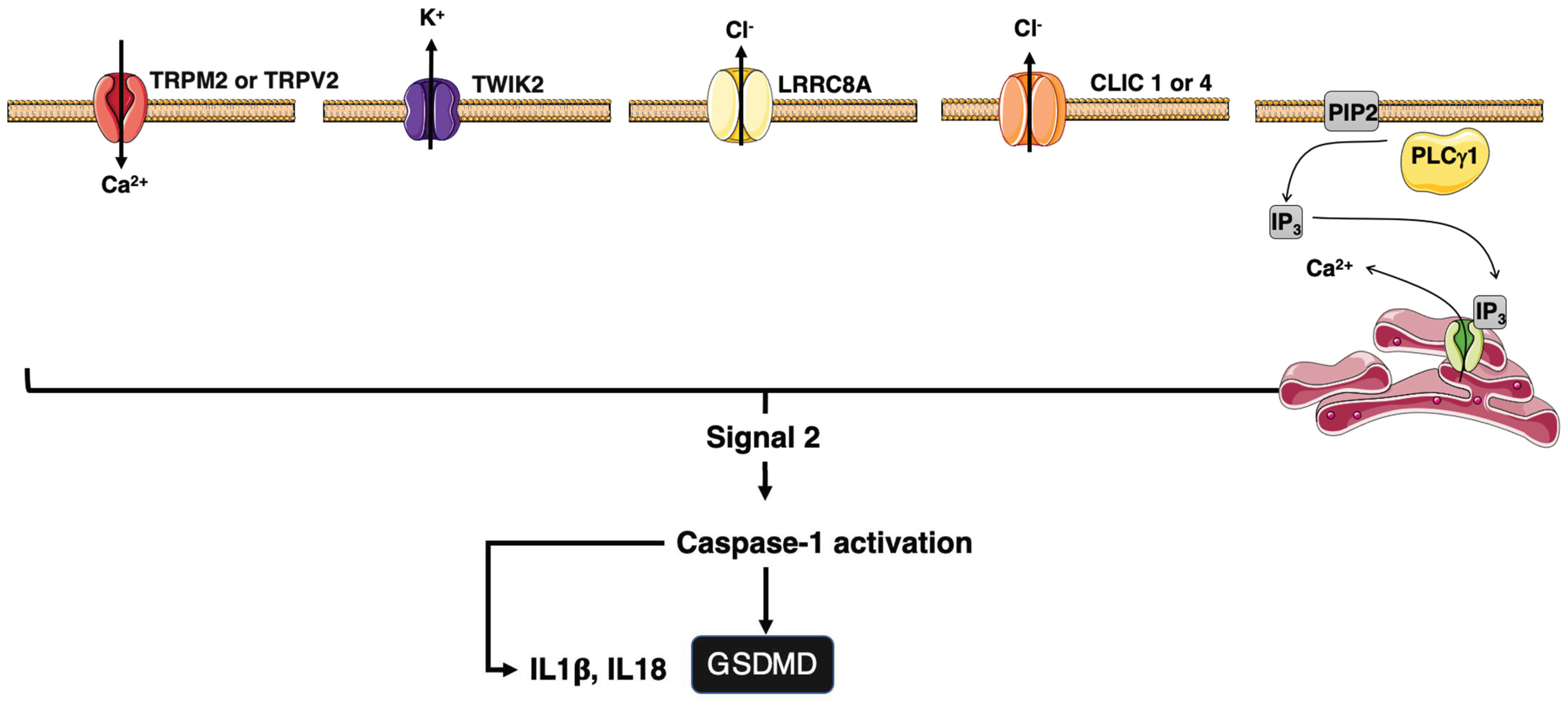
4.3. Ion Regulation of Inflammation
4.3.1. Ion Channels and Inflammasomes
4.3.2. Ion Channels and CD95
5. Conclusions and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kurma, K.; Boizard-Moracchini, A.; Galli, G.; Jean, M.; Vacher, P.; Blanco, P.; Legembre, P. Soluble CD95L in cancers and chronic inflammatory disorders, a new therapeutic target? Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188596. [Google Scholar] [CrossRef] [PubMed]
- Guegan, J.P.; Ginestier, C.; Charafe-Jauffret, E.; Ducret, T.; Quignard, J.F.; Vacher, P.; Legembre, P. CD95/Fas and metastatic disease: What does not kill you makes you stronger. Semin. Cancer Biol. 2019, 60, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Madkaikar, M.; Mhatre, S.; Gupta, M.; Ghosh, K. Advances in autoimmune lymphoproliferative syndromes. Eur. J. Haematol. 2011, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lenardo, M.J.; Oliveira, J.B.; Zheng, L.; Rao, V.K. ALPS-ten lessons from an international workshop on a genetic disease of apoptosis. Immunity 2010, 32, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.B.; Bleesing, J.J.; Dianzani, U.; Fleisher, T.A.; Jaffe, E.S.; Lenardo, M.J.; Rieux-Laucat, F.; Siegel, R.M.; Su, H.C.; Teachey, D.T.; et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood 2010, 116, e35–e40. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Huang, P.; Yang, Y.; Hao, M.; Peng, H.; Li, F. Updated Understanding of Autoimmune Lymphoproliferative Syndrome (ALPS). Clin. Rev. Allergy Immunol. 2016, 50, 55–63. [Google Scholar] [CrossRef]
- Fields, M.L.; Sokol, C.L.; Eaton-Bassiri, A.; Seo, S.; Madaio, M.P.; Erikson, J. Fas/Fas ligand deficiency results in altered localization of anti-double-stranded DNA B cells and dendritic cells. J. Immunol. 2001, 167, 2370–2378. [Google Scholar] [CrossRef] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297. [Google Scholar] [CrossRef] [Green Version]
- Krainer, J.; Siebenhandl, S.; Weinhausel, A. Systemic autoinflammatory diseases. J. Autoimmun. 2020, 109, 102421. [Google Scholar] [CrossRef] [PubMed]
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Tran Mau-Them, F.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis. 2017, 76, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286. [Google Scholar] [CrossRef]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef]
- Moghaddas, F.; Llamas, R.; De Nardo, D.; Martinez-Banaclocha, H.; Martinez-Garcia, J.J.; Mesa-Del-Castillo, P.; Baker, P.J.; Gargallo, V.; Mensa-Vilaro, A.; Canna, S.; et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann. Rheum. Dis. 2017, 76, 2085–2094. [Google Scholar] [CrossRef]
- Shoham, N.G.; Centola, M.; Mansfield, E.; Hull, K.M.; Wood, G.; Wise, C.A.; Kastner, D.L. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 13501–13506. [Google Scholar] [CrossRef] [Green Version]
- Tummers, B.; Mari, L.; Guy, C.S.; Heckmann, B.L.; Rodriguez, D.A.; Ruhl, S.; Moretti, J.; Crawford, J.C.; Fitzgerald, P.; Kanneganti, T.D.; et al. Caspase-8-Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 2020, 52, 994–1006.e8. [Google Scholar] [CrossRef]
- Siegel, R.M.; Frederiksen, J.K.; Zacharias, D.A.; Chan, F.K.-M.; Johnson, M.; Lynch, D.; Tsien, R.Y.; Lenardo, M.J. Fas Preassociation Required for Apoptosis Signaling and Dominant Inhibition by Pathogenic Mutations. Science 2000, 288, 2354–2357. [Google Scholar] [CrossRef] [Green Version]
- Papoff, G.; Cascino, I.; Eramo, A.; Starace, G.; Lynch, D.H.; Ruberti, G. An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J. Immunol. 1996, 156, 4622–4630. [Google Scholar] [PubMed]
- Cascino, I.; Papoff, G.; De Maria, R.; Testi, R.; Ruberti, G. Fas/Apo-1 (CD95) receptor lacking the intracytoplasmic signaling domain protects tumor cells from Fas-mediated apoptosis. J. Immunol. 1996, 156, 13–17. [Google Scholar] [PubMed]
- Cascino, I.; Fiucci, G.; Papoff, G.; Ruberti, G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J. Immunol. 1995, 154, 2706–2713. [Google Scholar]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem. 2006, 281, 13636–13643. [Google Scholar] [CrossRef] [Green Version]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Guegan, J.P.; Legembre, P. Nonapoptotic functions of Fas/CD95 in the immune response. FEBS J. 2018, 285, 809–827. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Fu, T.M.; Cruz, A.C.; Sengupta, P.; Thomas, S.K.; Wang, S.; Siegel, R.M.; Wu, H.; Chou, J.J. Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol. Cell 2016, 61, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, M.; Gunther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, F.; Zhang, X.; Zhang, H.; Zhao, Q.; Li, M.; Wu, X.; Wang, L.; Liu, J.; Wu, X.; et al. Caspase-8 auto-cleavage regulates programmed cell death and collaborates with RIPK3/MLKL to prevent lymphopenia. Cell Death Differ. 2022. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, B.D.; Leverrier, S.; Weist, B.M.; Newton, R.H.; Arechiga, A.F.; Luhrs, K.A.; Morrissette, N.S.; Walsh, C.M. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16677–16682. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Green, J.P.; Yu, S.; Martin-Sanchez, F.; Pelegrin, P.; Lopez-Castejon, G.; Lawrence, C.B.; Brough, D. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc. Natl. Acad. Sci. USA 2018, 115, E9371–E9380. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fan, C.; Zhang, H.; Zhao, Q.; Liu, Y.; Xu, C.; Xie, Q.; Wu, X.; Yu, X.; Zhang, J.; et al. MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 2016, 16, 3247–3259. [Google Scholar] [CrossRef] [Green Version]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-kappaB Subunits in the Control of Transcription. Cells 2016, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Udalova, I.A.; Richardson, A.; Denys, A.; Smith, C.; Ackerman, H.; Foxwell, B.; Kwiatkowski, D. Functional consequences of a polymorphism affecting NF-kappaB p50-p50 binding to the TNF promoter region. Mol. Cell Biol. 2000, 20, 9113–9119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I.; et al. KPC1-mediated ubiquitination and proteasomal processing of NF-kappaB1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.M.; Tran, A.C.; Grilli, M.; Lenardo, M.J. NF-kappa B subunit regulation in nontransformed CD4+ T lymphocytes. Science 1992, 256, 1452–1456. [Google Scholar] [CrossRef]
- Grundstrom, S.; Anderson, P.; Scheipers, P.; Sundstedt, A. Bcl-3 and NFkappaB p50-p50 homodimers act as transcriptional repressors in tolerant CD4+ T cells. J. Biol. Chem. 2004, 279, 8460–8468. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, B.C.; Legembre, P.; Pietras, E.; Bubici, C.; Franzoso, G.; Peter, M.E. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004, 23, 3175–3185. [Google Scholar] [CrossRef] [Green Version]
- Legembre, P.; Barnhart, B.C.; Peter, M.E. The relevance of NF-kappaB for CD95 signaling in tumor cells. Cell Cycle 2004, 3, 1235–1239. [Google Scholar] [CrossRef] [Green Version]
- Legembre, P.; Barnhart, B.C.; Zheng, L.; Vijayan, S.; Straus, S.E.; Puck, J.; Dale, J.K.; Lenardo, M.; Peter, M.E. Induction of apoptosis and activation of NF-kappaB by CD95 require different signalling thresholds. EMBO Rep. 2004, 5, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Hoogwater, F.J.; Nijkamp, M.W.; Smakman, N.; Steller, E.J.; Emmink, B.L.; Westendorp, B.F.; Raats, D.A.; Sprick, M.R.; Schaefer, U.; Van Houdt, W.J.; et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 2010, 138, 2357–2367. [Google Scholar] [CrossRef]
- Hoogwater, F.J.; Steller, E.J.; Westendorp, B.F.; Borel Rinkes, I.H.; Kranenburg, O. CD95 signaling in colorectal cancer. Biochim. Biophys. Acta 2012, 1826, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letellier, E.; Kumar, S.; Sancho-Martinez, I.; Krauth, S.; Funke-Kaiser, A.; Laudenklos, S.; Konecki, K.; Klussmann, S.; Corsini, N.S.; Kleber, S.; et al. CD95-ligand on peripheral myeloid cells activates Syk kinase to trigger their recruitment to the inflammatory site. Immunity 2010, 32, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malleter, M.; Tauzin, S.; Bessede, A.; Castellano, R.; Goubard, A.; Godey, F.; Leveque, J.; Jezequel, P.; Campion, L.; Campone, M.; et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res. 2013, 73, 6711–6721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steller, E.J.; Ritsma, L.; Raats, D.A.; Hoogwater, F.J.; Emmink, B.L.; Govaert, K.M.; Laoukili, J.; Rinkes, I.H.; van Rheenen, J.; Kranenburg, O. The death receptor CD95 activates the cofilin pathway to stimulate tumour cell invasion. EMBO Rep. 2011, 12, 931–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauzin, S.; Chaigne-Delalande, B.; Selva, E.; Khadra, N.; Daburon, S.; Contin-Bordes, C.; Blanco, P.; Le Seyec, J.; Ducret, T.; Counillon, L.; et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol. 2011, 9, e1001090. [Google Scholar] [CrossRef] [Green Version]
- Teodorczyk, M.; Kleber, S.; Wollny, D.; Sefrin, J.P.; Aykut, B.; Mateos, A.; Herhaus, P.; Sancho-Martinez, I.; Hill, O.; Gieffers, C.; et al. CD95 promotes metastatic spread via Sck in pancreatic ductal adenocarcinoma. Cell Death Differ. 2015, 22, 1192–1202. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Gulculer, G.S.; Golbach, L.; Block, H.; Zarbock, A.; Martin-Villalba, A. Endothelial cell-derived CD95 ligand serves as a chemokine in induction of neutrophil slow rolling and adhesion. Elife 2016, 5, e18542. [Google Scholar] [CrossRef]
- Poissonnier, A.; Guegan, J.P.; Nguyen, H.T.; Best, D.; Levoin, N.; Kozlov, G.; Gehring, K.; Pineau, R.; Jouan, F.; Morere, L.; et al. Disrupting the CD95-PLCgamma1 interaction prevents Th17-driven inflammation. Nat. Chem. Biol. 2018, 14, 1079–1089. [Google Scholar] [CrossRef]
- Poissonnier, A.; Sanseau, D.; Le Gallo, M.; Malleter, M.; Levoin, N.; Viel, R.; Morere, L.; Penna, A.; Blanco, P.; Dupuy, A.; et al. CD95-Mediated Calcium Signaling Promotes T Helper 17 Trafficking to Inflamed Organs in Lupus-Prone Mice. Immunity 2016, 45, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Guegan, J.P.; Pollet, J.; Ginestier, C.; Charafe-Jauffret, E.; Peter, M.E.; Legembre, P. CD95/Fas suppresses NF-kappaB activation through recruitment of KPC2 in a CD95L/FasL-independent mechanism. iScience 2021, 24, 103538. [Google Scholar] [CrossRef] [PubMed]
- Qadir, A.S.; Guegan, J.P.; Ginestier, C.; Chaibi, A.; Bessede, A.; Charafe-Jauffret, E.; Macario, M.; Lavoue, V.; Rouge, T.M.; Law, C.; et al. CD95/Fas protects triple negative breast cancer from anti-tumor activity of NK cells. iScience 2021, 24, 103348. [Google Scholar] [CrossRef] [PubMed]
- Rebe, C.; Cathelin, S.; Launay, S.; Filomenko, R.; Prevotat, L.; L’Ollivier, C.; Gyan, E.; Micheau, O.; Grant, S.; Dubart-Kupperschmitt, A.; et al. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood 2007, 109, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Dohrman, A.; Kataoka, T.; Cuenin, S.; Russell, J.Q.; Tschopp, J.; Budd, R.C. Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-kappa B activation. J. Immunol. 2005, 174, 5270–5278. [Google Scholar] [CrossRef] [Green Version]
- Alderson, M.R.; Armitage, R.J.; Maraskovsky, E.; Tough, T.W.; Roux, E.; Schooley, K.; Ramsdell, F.; Lynch, D.H. Fas transduces activation signals in normal human T lymphocytes. J. Exp. Med. 1993, 178, 2231–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderson, M.R.; Tough, T.W.; Braddy, S.; Davis-Smith, T.; Roux, E.; Schooley, K.; Miller, R.E.; Lynch, D.H. Regulation of apoptosis and T cell activation by Fas-specific mAb. Int. Immunol. 1994, 6, 1799–1806. [Google Scholar] [CrossRef]
- Parry, R.; Smith, G.; Reif, K.; Sansom, D.M.; Ward, S. Activation of the PI3K effector protein kinase B following ligation of CD28 or Fas. Biochem. Soc. Trans. 1997, 25, S589. [Google Scholar] [CrossRef] [Green Version]
- Gulbins, E.; Brenner, B.; Koppenhoefer, U.; Linderkamp, O.; Lang, F. Fas or ceramide induce apoptosis by Ras-regulated phosphoinositide-3-kinase activation. J. Leukoc. Biol. 1998, 63, 253–263. [Google Scholar] [CrossRef]
- Gulbins, E.; Hermisson, M.; Brenner, B.; Grassme, H.U.; Linderkamp, O.; Dichgans, J.; Weller, M.; Lang, F. Cellular stimulation via CD95 involves activation of phospho-inositide-3-kinase. Pflug. Arch. 1998, 435, 546–554. [Google Scholar] [CrossRef]
- Fujita, E.; Kouroku, Y.; Miho, Y.; Tsukahara, T.; Ishiura, S.; Momoi, T. Wortmannin enhances activation of CPP32 (Caspase-3) induced by TNF or anti-Fas. Cell Death Differ. 1998, 5, 289–297. [Google Scholar] [CrossRef]
- Cursi, S.; Rufini, A.; Stagni, V.; Condo, I.; Matafora, V.; Bachi, A.; Bonifazi, A.P.; Coppola, L.; Superti-Furga, G.; Testi, R.; et al. Src kinase phosphorylates Caspase-8 on Tyr380: A novel mechanism of apoptosis suppression. EMBO J. 2006, 25, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Senft, J.; Helfer, B.; Frisch, S.M. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 2007, 67, 11505–11509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinehr, R.; Schliess, F.; Haussinger, D. Hyperosmolarity and CD95L trigger CD95/EGF receptor association and tyrosine phosphorylation of CD95 as prerequisites for CD95 membrane trafficking and DISC formation. FASEB J. 2003, 17, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Jin, C.; Flavell, R.A. Inflammasome activation. The missing link: How the inflammasome senses oxidative stress. Immunol. Cell Biol. 2010, 88, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [PubMed]
- Mouasni, S.; Gonzalez, V.; Schmitt, A.; Bennana, E.; Guillonneau, F.; Mistou, S.; Avouac, J.; Ea, H.K.; Devauchelle, V.; Gottenberg, J.E.; et al. The classical NLRP3 inflammasome controls FADD unconventional secretion through microvesicle shedding. Cell Death Dis. 2019, 10, 190. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Seino, K.; Kayagaki, N.; Okumura, K.; Yagita, H. Antitumor effect of locally produced CD95 ligand. Nat. Med. 1997, 3, 165–170. [Google Scholar] [CrossRef]
- Miwa, K.; Asano, M.; Horai, R.; Iwakura, Y.; Nagata, S.; Suda, T. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med. 1998, 4, 1287–1292. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kayagaki, N.; Kuida, K.; Nakano, H.; Hayashi, N.; Takeda, K.; Matsui, K.; Kashiwamura, S.; Hada, T.; Akira, S.; et al. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 1999, 11, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geserick, P.; Hupe, M.; Moulin, M.; Wong, W.W.; Feoktistova, M.; Kellert, B.; Gollnick, H.; Silke, J.; Leverkus, M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 2009, 187, 1037–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Juliana, C.; Solorzano, L.; Kang, S.; Wu, J.; Datta, P.; McCormick, M.; Huang, L.; McDermott, E.; et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010, 11, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef]
- Roth, S.; Cao, J.; Singh, V.; Tiedt, S.; Hundeshagen, G.; Li, T.; Boehme, J.D.; Chauhan, D.; Zhu, J.; Ricci, A.; et al. Post-injury immunosuppression and secondary infections are caused by an AIM2 inflammasome-driven signaling cascade. Immunity 2021, 54, 648–659.e8. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taieb, J.; Chaput, N.; Menard, C.; Apetoh, L.; Ullrich, E.; Bonmort, M.; Pequignot, M.; Casares, N.; Terme, M.; Flament, C.; et al. A novel dendritic cell subset involved in tumor immunosurveillance. Nat. Med. 2006, 12, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; McGowan, J.; Antonovitch, J.; Gao, K.M.; Jiang, Z.; Sharma, S.; Baltus, G.A.; Nickerson, K.M.; Marshak-Rothstein, A.; Fitzgerald, K.A. cGAS-STING Pathway Does Not Promote Autoimmunity in Murine Models of SLE. Front. Immunol. 2021, 12, 605930. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.R.; Kozyrev, S.V.; Baechler, E.C.; Reddy, M.V.; Plenge, R.M.; Bauer, J.W.; Ortmann, W.A.; Koeuth, T.; Gonzalez Escribano, M.F.; Argentine and Spanish Collaborative Groups; et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat. Genet. 2006, 38, 550–555. [Google Scholar] [CrossRef]
- Song, S.; De, S.; Nelson, V.; Chopra, S.; LaPan, M.; Kampta, K.; Sun, S.; He, M.; Thompson, C.D.; Li, D.; et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J. Clin. Invest. 2020, 130, 6700–6717. [Google Scholar] [CrossRef]
- Tada, Y.; Kondo, S.; Aoki, S.; Koarada, S.; Inoue, H.; Suematsu, R.; Ohta, A.; Mak, T.W.; Nagasawa, K. Interferon regulatory factor 5 is critical for the development of lupus in MRL/lpr mice. Arthritis Rheumatol. 2011, 63, 738–748. [Google Scholar] [CrossRef]
- Ip, W.K.; Medzhitov, R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 2015, 6, 6931. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Compan, V.; Baroja-Mazo, A.; Lopez-Castejon, G.; Gomez, A.I.; Martinez, C.M.; Angosto, D.; Montero, M.T.; Herranz, A.S.; Bazan, E.; Reimers, D.; et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 2012, 37, 487–500. [Google Scholar] [CrossRef] [Green Version]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e54. [Google Scholar] [CrossRef] [Green Version]
- Jentsch, T.J. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Green, J.P.; Swanton, T.; Morris, L.V.; El-Sharkawy, L.Y.; Cook, J.; Yu, S.; Beswick, J.; Adamson, A.D.; Humphreys, N.E.; Bryce, R.; et al. LRRC8A is essential for hypotonicity-, but not for DAMP-induced NLRP3 inflammasome activation. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Argenzio, E.; Moolenaar, W.H. Emerging biological roles of Cl- intracellular channel proteins. J. Cell Sci. 2016, 129, 4165–4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo-Fernandez, R.; Coll, R.C.; Kearney, J.; Breit, S.; O’Neill, L.A.J. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017, 292, 12077–12087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef]
- Shimizu, T.; Numata, T.; Okada, Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(-) channel. Proc. Natl. Acad. Sci. USA 2004, 101, 6770–6773. [Google Scholar] [CrossRef] [Green Version]
- Lepple-Wienhues, A.; Szabo, I.; Laun, T.; Kaba, N.K.; Gulbins, E.; Lang, F. The tyrosine kinase p56lck mediates activation of swelling-induced chloride channels in lymphocytes. J. Cell Biol. 1998, 141, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Szabo, I.; Lepple-Wienhues, A.; Kaba, K.N.; Zoratti, M.; Gulbins, E.; Lang, F. Tyrosine kinase-dependent activation of a chloride channel in CD95-induced apoptosis in T lymphocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 6169–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulbins, E.; Szabo, I.; Baltzer, K.; Lang, F. Ceramide-induced inhibition of T lymphocyte voltage-gated potassium channel is mediated by tyrosine kinases. Proc. Natl. Acad. Sci. USA 1997, 94, 7661–7666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, I.; Gulbins, E.; Apfel, H.; Zhang, X.; Barth, P.; Busch, A.E.; Schlottmann, K.; Pongs, O.; Lang, F. Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J. Biol. Chem. 1996, 271, 20465–20469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Khadra, N.; Bresson-Bepoldin, L.; Penna, A.; Chaigne-Delalande, B.; Segui, B.; Levade, T.; Vacher, A.M.; Reiffers, J.; Ducret, T.; Moreau, J.F.; et al. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) beta2, and prevents death-inducing signaling complex formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19072–19077. [Google Scholar] [CrossRef] [Green Version]
- Quistad, S.D.; Traylor-Knowles, N. Precambrian origins of the TNFR superfamily. Cell Death Discov. 2016, 2, 16058. [Google Scholar] [CrossRef]
- Huang, S.; Wang, X.; Yan, Q.; Guo, L.; Yuan, S.; Huang, G.; Huang, H.; Li, J.; Dong, M.; Chen, S.; et al. The evolution and regulation of the mucosal immune complexity in the basal chordate amphioxus. J. Immunol. 2011, 186, 2042–2055. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| ALPS Type | Mutation Type |
|---|---|
| ALPS-FAS | CD95 germline homo- or heterozygous mutation |
| ALPS-sFAS | CD95 somatic mutation |
| ALPS-FASLG | CD95L germline mutation |
| ALPS-CASP10 | Caspase 10 germline mutations |
| ALPS-U | ALPS phenotype with no known-ALPS mutation |
| DAMP-Sensor Mutation | Mutation Type | Autoinflammatory Disorder |
|---|---|---|
| NLRP1 | Loss of function | NAIAD |
| NLRP3 | Gain of function | CAPS = NOMID, FCAS, MWS |
| NLRC4 | Gain of function | SCAN4, MAS |
| Pyrin | Gain of function of MEFV or loss of function of MVK | FMF, HIDS, PAAND |
| PSTPIP1 | Loss of function | PAPA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galli, G.; Vacher, P.; Ryffel, B.; Blanco, P.; Legembre, P. Fas/CD95 Signaling Pathway in Damage-Associated Molecular Pattern (DAMP)-Sensing Receptors. Cells 2022, 11, 1438. https://doi.org/10.3390/cells11091438
Galli G, Vacher P, Ryffel B, Blanco P, Legembre P. Fas/CD95 Signaling Pathway in Damage-Associated Molecular Pattern (DAMP)-Sensing Receptors. Cells. 2022; 11(9):1438. https://doi.org/10.3390/cells11091438
Chicago/Turabian StyleGalli, Gael, Pierre Vacher, Bernhard Ryffel, Patrick Blanco, and Patrick Legembre. 2022. "Fas/CD95 Signaling Pathway in Damage-Associated Molecular Pattern (DAMP)-Sensing Receptors" Cells 11, no. 9: 1438. https://doi.org/10.3390/cells11091438
APA StyleGalli, G., Vacher, P., Ryffel, B., Blanco, P., & Legembre, P. (2022). Fas/CD95 Signaling Pathway in Damage-Associated Molecular Pattern (DAMP)-Sensing Receptors. Cells, 11(9), 1438. https://doi.org/10.3390/cells11091438