
A Comprehensive Investigation of Steroidogenic Signaling in Classical and New Experimental Cell Models of Adrenocortical Carcinoma

 , , , ,
, , , ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cancer Cell Lines
2.2. Whole Genome Sequencing and Genomic Data Presentation
2.3. Stimulation Experiments
2.4. Quantitative Real-Time PCR
2.5. Liquid Chromatography Tandem Mass Spectrometry (LC–MS/MS) Steroid Measurements
2.6. Protein Quantification
2.7. Electrophysiological Studies
2.8. Statistical Analysis and Graphical Designs
3. Results
3.1. Establishment of a Novel Cancer Cell Line, TVBF-7
3.2. Mutational Status of Important Driver Genes
3.3. Baseline Gene Expression Levels and Electrophysiological Properties
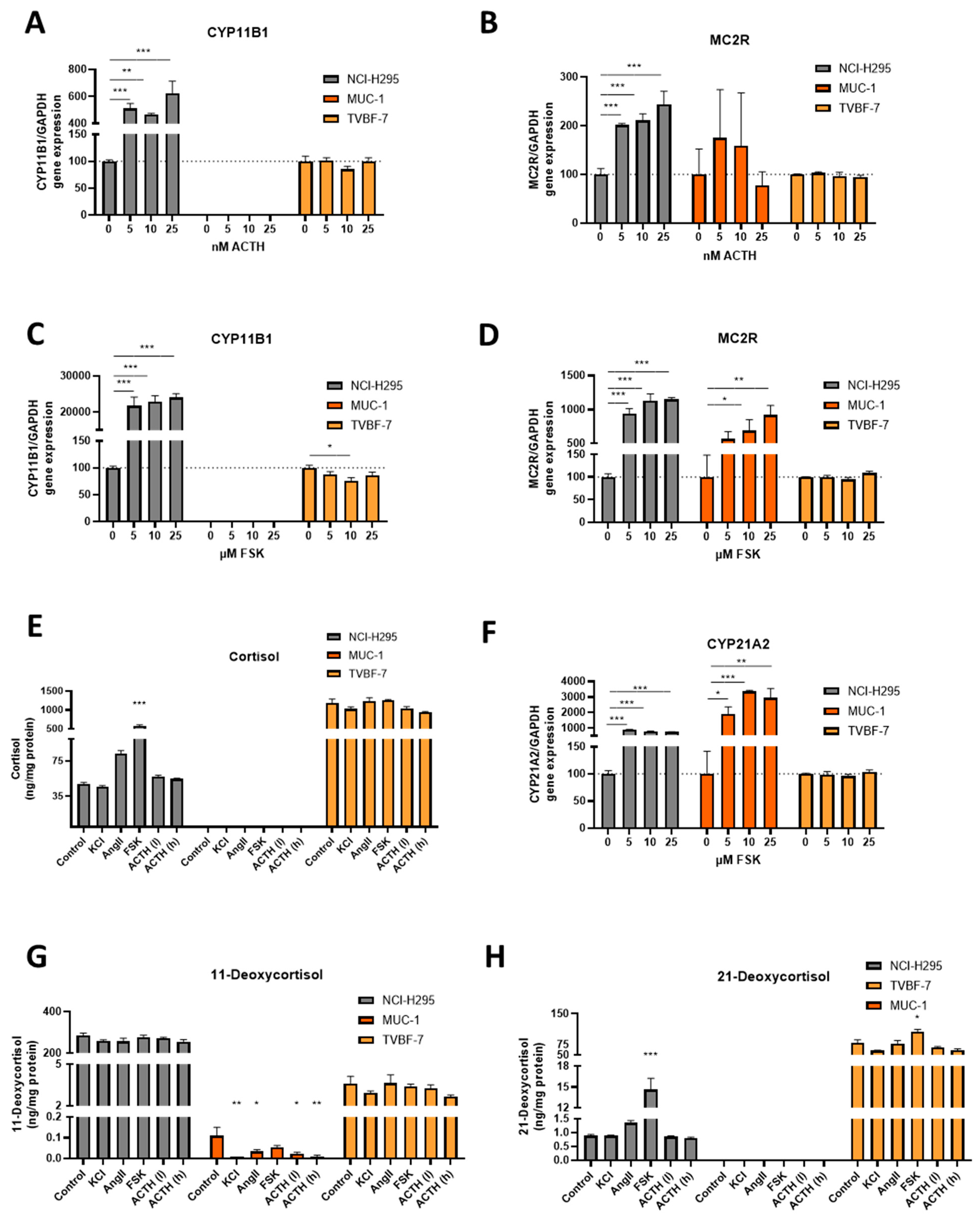
3.4. Stimulation of the First Steps of Steroidogenesis upon Known Stimuli: TVBF-7 Are Unresponsive
3.5. Mineralocorticoid Pathway Stimulation Reveals Dysregulated and Distinct Patterns among the Cell Lines
3.6. Responsive, Non-Producing and Autonomous Secretion Detected Regarding the Glucocorticoid Production
3.7. MUC-1 Induce Androgen Production and Upregulation of the AR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fassnacht, M.; Dekkers, O.M.; Else, T.; Baudin, E.; Berruti, A.; de Krijger, R.; Haak, H.R.; Mihai, R.; Assie, G.; Terzolo, M. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2018, 179, G1–G46. [Google Scholar] [CrossRef]
- Schweitzer, S.; Kunz, M.; Kurlbaum, M.; Vey, J.; Kendl, S.; Deutschbein, T.; Hahner, S.; Fassnacht, M.; Dandekar, T.; Kroiss, M. Plasma steroid metabolome profiling for the diagnosis of adrenocortical carcinoma. Eur. J. Endocrinol. 2019, 180, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Arlt, W.; Biehl, M.; Taylor, A.E.; Hahner, S.; Libe, R.; Hughes, B.A.; Schneider, P.; Smith, D.J.; Stiekema, H.; Krone, N.; et al. Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. J. Clin. Endocrinol. Metab. 2011, 96, 3775–3784. [Google Scholar] [CrossRef] [PubMed]
- Bancos, I.; Taylor, A.E.; Chortis, V.; Sitch, A.J.; Jenkinson, C.; Davidge-Pitts, C.J.; Lang, K.; Tsagarakis, S.; Macech, M.; Riester, A.; et al. Urine steroid metabolomics for the differential diagnosis of adrenal incidentalomas in the EURINE-ACT study: A prospective test validation study. Lancet Diabetes Endocrinol. 2020, 8, 773–781. [Google Scholar] [CrossRef]
- Chortis, V.; Bancos, I.; Nijman, T.; Gilligan, L.C.; Taylor, A.E.; Ronchi, C.L.; O’reilly, M.W.; Schreiner, J.; Asia, M.; Riester, A.; et al. Urine Steroid Metabolomics as a Novel Tool for Detection of Recurrent Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2020, 105, e307–e318. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Chang, Y.H.; Pu, H.F. Mitotane exhibits dual effects on steroidogenic enzymes gene transcription under basal and cAMP-stimulating microenvironments in NCI-H295 cells. Toxicology 2012, 298, 14–23. [Google Scholar] [CrossRef]
- Lehmann, T.P.; Wrzesinski, T.; Jagodzinski, P.P. The effect of mitotane on viability, steroidogenesis and gene expression in NCIH295R adrenocortical cells. Mol. Med. Rep. 2013, 7, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Minamidate, T.; Shiga, A.; Ruike, Y.; Ishiwata, K.; Naito, K.; Ishida, A.; Deguchi, H.; Fujimoto, M.; Koide, H.; et al. Steroid metabolites for diagnosing and predicting clinicopathological features in cortisol-producing adrenocortical carcinoma. BMC Endocr. Disord. 2020, 20, 173. [Google Scholar] [CrossRef] [PubMed]
- Abiven, G.; Coste, J.; Groussin, L.; Anract, P.; Tissier, F.; Legmann, P.; Dousset, B.; Bertagna, X.; Bertherat, J. Clinical and biological features in the prognosis of adrenocortical cancer: Poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J. Clin. Endocrinol. Metab. 2006, 91, 2650–2655. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, M.L.; Curlee, K.; Lloyd, R.; Farley, D.R.; Grant, C.S.; Thompson, G.B.; Rowland, C.; Young, W.F.; van Heerden, J. Aldosterone-secreting adrenocortical carcinomas are associated with unique operative risks and outcomes. Surgery 2002, 132, 1008–1011; discussion 1012. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Bonafe, M.; Bravaccini, S. Androgen receptor in breast cancer: A wolf in sheep’s clothing? A lesson from prostate cancer. Semin. Cancer Biol. 2020, 60, 132–137. [Google Scholar] [CrossRef]
- Snaterse, G.; Visser, J.A.; Arlt, W.; Hofland, J. Circulating steroid hormone variations throughout different stages of prostate cancer. Endocr. Relat. Cancer 2017, 24, R403–R420. [Google Scholar] [CrossRef]
- Pinto, E.M.; Kiseljak-Vassiliades, K.; Hantel, C. Contemporary preclinical human models of adrenocortical carcinoma. Curr. Opin. Endocr. Metab. Res. 2019, 8, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Oie, H.K.; Shackleton, C.H.; Chen, T.R.; Triche, T.J.; Myers, C.E.; Chrousos, G.P.; Brennan, M.F.; Stein, C.A.; La Rocca, R.V. Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis. Cancer Res. 1990, 50, 5488–5496. [Google Scholar]
- Hantel, C.; Shapiro, I.; Poli, G.; Chiapponi, C.; Bidlingmaier, M.; Reincke, M.; Luconi, M.; Jung, S.; Beuschlein, F. Targeting heterogeneity of adrenocortical carcinoma: Evaluation and extension of preclinical tumor models to improve clinical translation. Oncotarget 2016, 7, 79292–79304. [Google Scholar] [CrossRef] [PubMed]
- Kiseljak-Vassiliades, K.; Zhang, Y.; Bagby, S.M.; Kar, A.; Pozdeyev, N.; Xu, M.; Gowan, K.; Sharma, V.; Raeburn, C.D.; Albuja-Cruz, M.; et al. Development of new preclinical models to advance adrenocortical carcinoma research. Endocr. Relat. Cancer 2018, 25, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Landwehr, L.S.; Schreiner, J.; Appenzeller, S.; Kircher, S.; Herterich, S.; Sbiera, S.; Fassnacht, M.; Kroiss, M.; Weigand, I. A novel patient-derived cell line of adrenocortical carcinoma shows a pathogenic role of germline MUTYH mutation and high tumour mutational burden. Eur. J. Endocrinol. 2021, 184, 823–835. [Google Scholar] [CrossRef]
- Vicennati, V.; Repaci, A.; di Dalmazi, G.; Rinaldi, E.; Golfieri, R.; Giampalma, E.; Minni, F.; Marrano, N.; Santini, D.; Pasquali, R. Combined aldosterone and cortisol secretion by adrenal incidentaloma. Int. J. Surg. Pathol. 2012, 20, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef]
- Bothou, C.; Sharma, A.; Oo, A.; Kim, B.; Perge, P.; Igaz, P.; Ronchi, C.L.; Shapiro, I.; Hantel, C. Novel Insights into the Molecular Regulation of Ribonucleotide Reductase in Adrenocortical Carcinoma Treatment. Cancers 2021, 13, 4200. [Google Scholar] [CrossRef] [PubMed]
- Warde, K.M.; Schoenmakers, E.; Ribes Martinez, E.; Lim, Y.J.; Leonard, M.; Lawless, S.J.; O’Shea, P.; Chatterjee, V.K.; Gurnell, M.; Hantel, C.; et al. Liver X receptor inhibition potentiates mitotane-induced adrenotoxicity in ACC. Endocr. Relat. Cancer 2020, 27, 361–373. [Google Scholar] [CrossRef]
- Siebert, C.; Ciato, D.; Murakami, M.; Frei-Stuber, L.; Perez-Rivas, L.G.; Monteserin-Garcia, J.L.; Nölting, S.; Maurer, J.; Feuchtinger, A.; Walch, A.K.; et al. Heat Shock Protein 90 as a Prognostic Marker and Therapeutic Target for Adrenocortical Carcinoma. Front. Endocrinol. 2019, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Hasanovic, A.; Ruggiero, C.; Jung, S.; Rapa, I.; Signetti, L.; Ben Hadj, M.; Terzolo, M.; Beuschlein, F.; Volante, M.; Hantel, C.; et al. Targeting the multidrug transporter Patched potentiates chemotherapy efficiency on adrenocortical carcinoma in vitro and in vivo. Int. J. Cancer 2018, 143, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Fragni, M.; Palma Lopez, L.P.; Rossini, E.; Abate, A.; Cosentini, D.; Salvi, V.; Vezzoli, S.; Poliani, P.L.; Bosisio, D.; Hantel, C.; et al. In Vitro cytotoxicity of cabazitaxel in adrenocortical carcinoma cell lines and human adrenocortical carcinoma primary cell cultures. Mol. Cell Endocrinol. 2019, 498, 110585. [Google Scholar] [CrossRef]
- Liang, R.; Weigand, I.; Lippert, J.; Kircher, S.; Altieri, B.; Steinhauer, S.; Hantel, C.; Rost, S.; Rosenwald, A.; Kroiss, M.; et al. Targeted Gene Expression Profile Reveals CDK4 as Therapeutic Target for Selected Patients With Adrenocortical Carcinoma. Front. Endocrinol. 2020, 11, 219. [Google Scholar] [CrossRef]
- Abate, A.; Rossini, E.; Bonini, S.A.; Fragni, M.; Cosentini, D.; Tiberio, G.A.M.; Benetti, D.; Hantel, C.; Laganà, M.; Grisanti, S.; et al. Cytotoxic Effect of Trabectedin in Human Adrenocortical Carcinoma Cell Lines and Primary Cells. Cancers 2020, 12, 928. [Google Scholar] [CrossRef]
- Rossini, E.; Tamburello, M.; Abate, A.; Beretta, S.; Fragni, M.; Cominelli, M.; Cosentini, D.; Hantel, C.; Bono, F.; Grisanti, S.; et al. Cytotoxic Effect of Progesterone, Tamoxifen and Their Combination in Experimental Cell Models of Human Adrenocortical Cancer. Front. Endocrinol. 2021, 12, 669426. [Google Scholar] [CrossRef] [PubMed]
- Cantini, G.; Fei, L.; Canu, L.; Lazzeri, E.; Sottili, M.; Francalanci, M.; Angelotti, M.L.; De Filpo, G.; Ercolino, T.; Gelmini, S.; et al. Stimulated Expression of CXCL12 in Adrenocortical Carcinoma by the PPARgamma Ligand Rosiglitazone Impairs Cancer Progression. J. Pers. Med. 2021, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.; Rossini, E.; Tamburello, M.; Lagana, M.; Cosentini, D.; Grisanti, S.; Fiorentini, C.; Tiberio, G.A.; Scatolini, M.; Grosso, E.; et al. Ribociclib Cytotoxicity Alone or Combined with Progesterone and/or Mitotane in In Vitro Adrenocortical Carcinoma Cells. Endocrinology 2022, 163, bqab248. [Google Scholar] [CrossRef]
- Hosogi, H.; Nagayama, S.; Kanamoto, N.; Yoshizawa, A.; Suzuki, T.; Nakao, K.; Sakai, Y. Biallelic APC inactivation was responsible for functional adrenocortical adenoma in familial adenomatous polyposis with novel germline mutation of the APC gene: Report of a case. Jpn. J. Clin. Oncol. 2009, 39, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Altieri, B.; Ronchi, C.L.; Kroiss, M.; Fassnacht, M. Next-generation therapies for adrenocortical carcinoma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101434. [Google Scholar] [CrossRef] [PubMed]
- Gara, S.K.; Lack, J.; Zhang, L.; Harris, E.; Cam, M.; Kebebew, E. Metastatic adrenocortical carcinoma displays higher mutation rate and tumor heterogeneity than primary tumors. Nat. Commun. 2018, 9, 4172. [Google Scholar] [CrossRef]
- Dierks, A.; Lichtenauer, U.D.; Sackmann, S.; Spyroglou, A.; Shapiro, I.; Geyer, M.; Manonopoulou, J.; Reincke, M.; Hantel, C.; Beuschlein, F. Identification of adrenal genes regulated in a potassium-dependent manner. J. Mol. Endocrinol. 2010, 45, 193–206. [Google Scholar] [CrossRef]
- Ueland, G.A.; Grinde, T.; Methlie, P.; Kelp, O.; Lovas, K.; Husebye, E.S. Diagnostic testing of autonomous cortisol secretion in adrenal incidentalomas. Endocr. Connect. 2020, 9, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, S.; Pinson, S.; Gimenez-Roqueplo, A.P.; Amar, L.; Ragazzon, B.; Launay, P.; Meatchi, T.; Libé, R.; Bertagna, X.; Audebourg, A.; et al. Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin. Cancer Res. 2010, 16, 5133–5141. [Google Scholar] [CrossRef]
- Assie, G.; Letouze, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; René-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Audenet, F.; Mejean, A.; Chartier-Kastler, E.; Roupret, M. Adrenal tumours are more predominant in females regardless of their histological subtype: A review. World J. Urol. 2013, 31, 1037–1043. [Google Scholar] [CrossRef]
- Alesina, P.F.; Walz, M.K. Adrenal Tumors: Are Gender Aspects Relevant? Visc. Med. 2020, 36, 15–19. [Google Scholar] [CrossRef]
- Sekido, R.; Lovell-Badge, R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 2008, 453, 930–934. [Google Scholar] [CrossRef]
- Ganguly, S.; Naik, D.; Muskara, A.; Mian, O.Y. The Nexus of Endocrine Signaling and Cancer: How Steroid Hormones Influence Genomic Stability. Endocrinology 2021, 162, bqaa177. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.F.; Mo, Q.; Hu, S.; Garippa, C.; Simon, N.G. Dehydroepiandrosterone upregulates neural androgen receptor level and transcriptional activity. J. Neurobiol. 2003, 57, 163–171. [Google Scholar] [CrossRef]
- Mo, Q.; Lu, S.F.; Hu, S.; Simon, N.G. DHEA and DHEA sulfate differentially regulate neural androgen receptor and its transcriptional activity. Brain Res. Mol. Brain Res. 2004, 126, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.K.; Berk, M.; Chung, Y.M.; Willard, B.; Bareja, R.; Rubin, M.; Sboner, A.; Sharifi, N. Loss of an Androgen-Inactivating and Isoform-Specific HSD17B4 Splice form Enables Emergence of Castration-Resistant Prostate Cancer. Cell Rep. 2018, 22, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Violante, S.; Achetib, N.; van Roermund, C.W.T.; Hagen, J.; Dodatko, T.; Vaz, F.M.; Waterham, H.R.; Chen, H.; Baes, M.; Yu, C.; et al. Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 2019, 33, 4355–4364. [Google Scholar] [CrossRef]
- Warde, K.; Lim, Y.-J.; Beuschlein, F.; Hantel, C.; Dennedy, M.C. Investigating the Role of Cholesterol and Lipid Trafficking in Mitotane Resistance in Adrenocortical Carcinoma. J. Endocr. Soc. 2021, 5 (Suppl. 1), A70. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sigala, S.; Bothou, C.; Penton, D.; Abate, A.; Peitzsch, M.; Cosentini, D.; Tiberio, G.A.M.; Bornstein, S.R.; Berruti, A.; Hantel, C. A Comprehensive Investigation of Steroidogenic Signaling in Classical and New Experimental Cell Models of Adrenocortical Carcinoma. Cells 2022, 11, 1439. https://doi.org/10.3390/cells11091439
Sigala S, Bothou C, Penton D, Abate A, Peitzsch M, Cosentini D, Tiberio GAM, Bornstein SR, Berruti A, Hantel C. A Comprehensive Investigation of Steroidogenic Signaling in Classical and New Experimental Cell Models of Adrenocortical Carcinoma. Cells. 2022; 11(9):1439. https://doi.org/10.3390/cells11091439
Chicago/Turabian StyleSigala, Sandra, Christina Bothou, David Penton, Andrea Abate, Mirko Peitzsch, Deborah Cosentini, Guido A. M. Tiberio, Stefan R. Bornstein, Alfredo Berruti, and Constanze Hantel. 2022. "A Comprehensive Investigation of Steroidogenic Signaling in Classical and New Experimental Cell Models of Adrenocortical Carcinoma" Cells 11, no. 9: 1439. https://doi.org/10.3390/cells11091439
APA StyleSigala, S., Bothou, C., Penton, D., Abate, A., Peitzsch, M., Cosentini, D., Tiberio, G. A. M., Bornstein, S. R., Berruti, A., & Hantel, C. (2022). A Comprehensive Investigation of Steroidogenic Signaling in Classical and New Experimental Cell Models of Adrenocortical Carcinoma. Cells, 11(9), 1439. https://doi.org/10.3390/cells11091439