
Signal Transduction Regulators in Axonal Regeneration



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
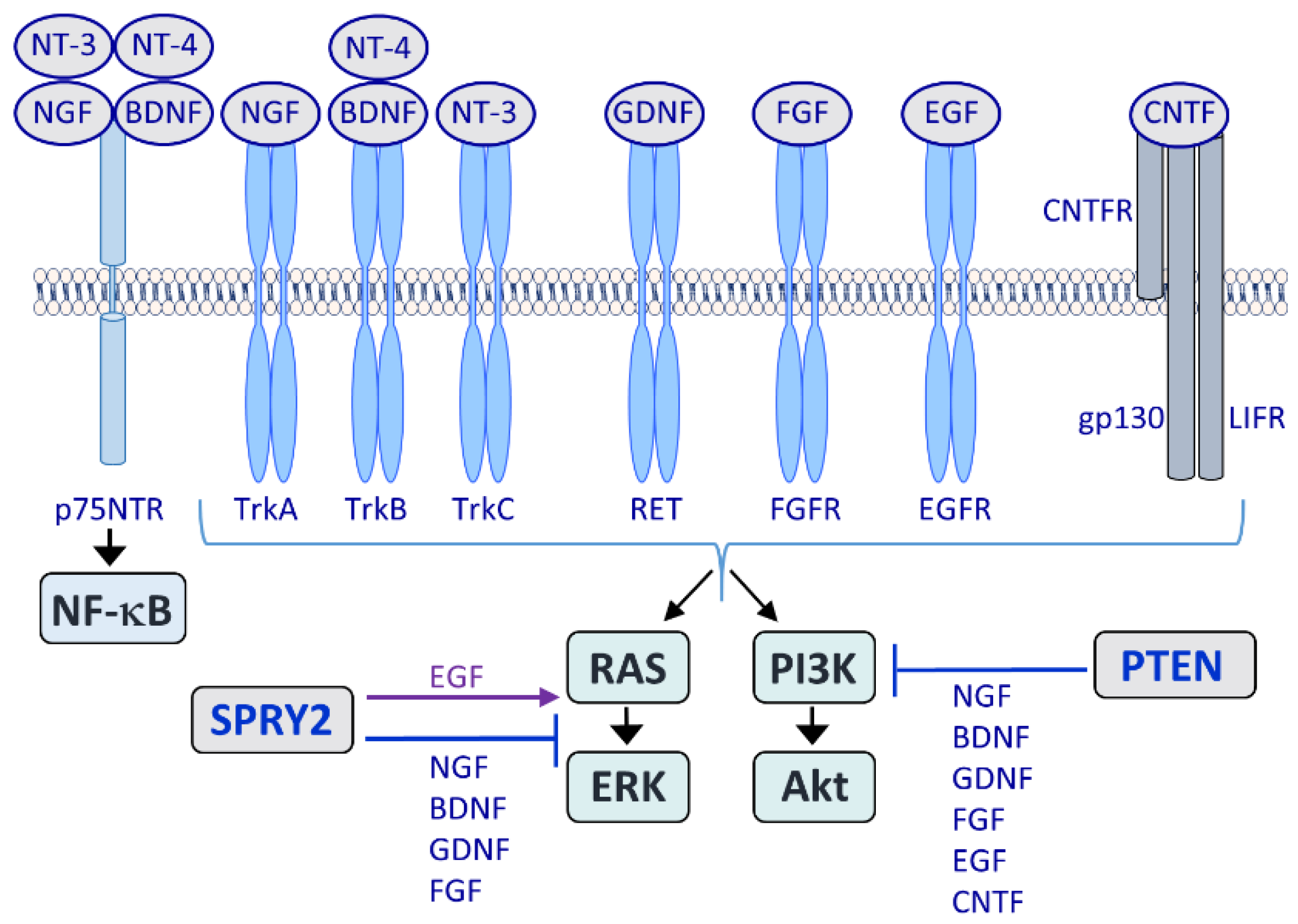
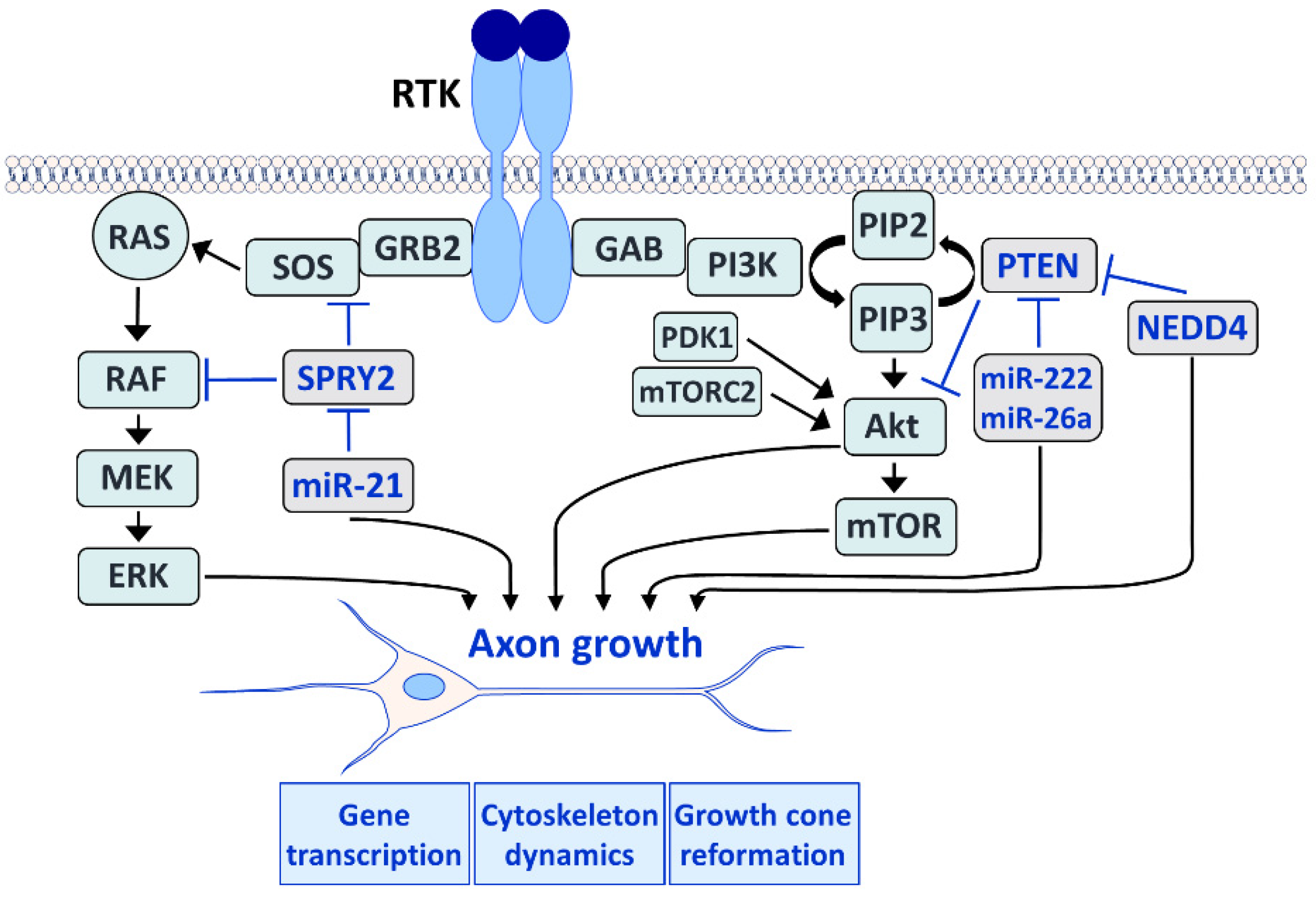
2. The Role of RAS/ERK and PI3K/Akt Signaling in Axonal Regeneration
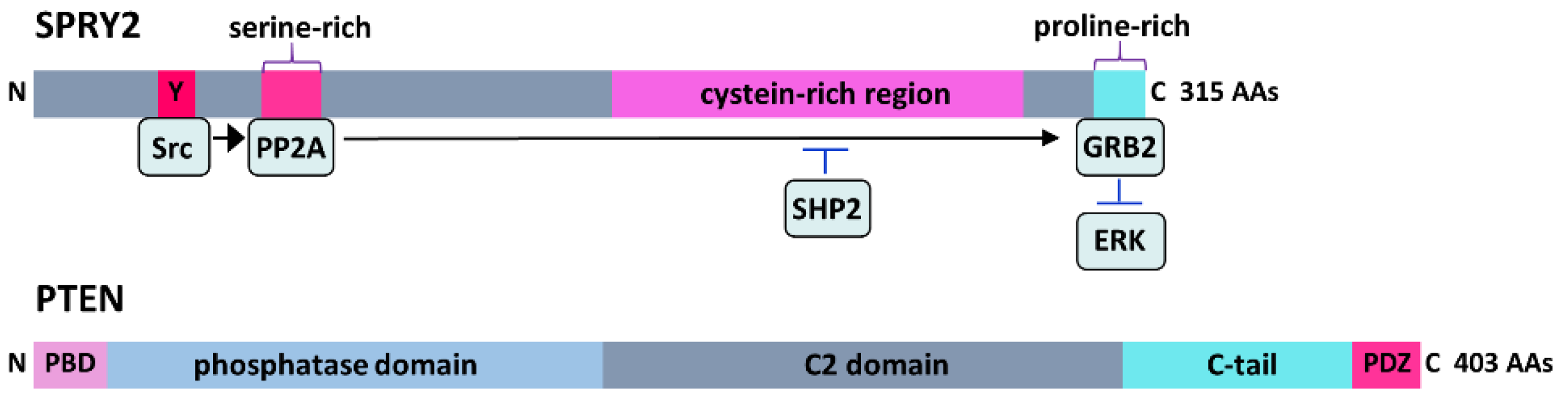
3. SPRY: Signal Regulators of RAS/ERK Signaling
3.1. SPRY and Development
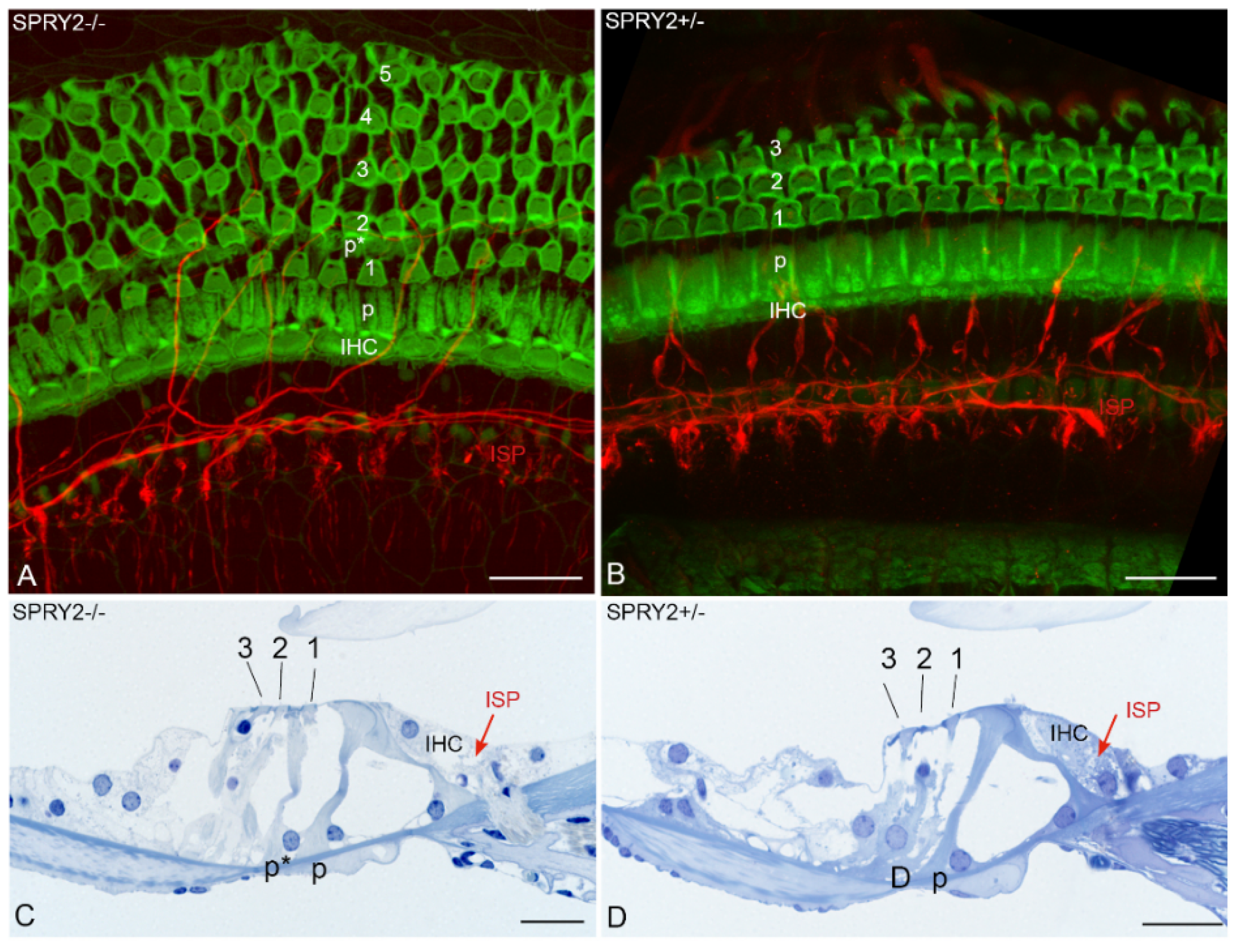
3.2. SPRY and Axonal Regeneration
4. PTEN: Signal Regulator of the PI3K/Akt Pathway
4.1. PTEN and Development
4.2. PTEN and Axonal Regeneration
5. Combined Approaches to Enhance Axonal Regeneration
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Chan, K.M.; Gordon, T.; Zochodne, D.W.; Power, H.A. Improving peripheral nerve regeneration: From molecular mechanisms to potential therapeutic targets. Exp. Neurol. 2014, 261, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Mar, F.M.; Bonni, A.; Sousa, M.M. Cell intrinsic control of axon regeneration. EMBO Rep. 2014, 15, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagappan, P.G.; Chen, H.; Wang, D.Y. Neuroregeneration and plasticity: A review of the physiological mechanisms for achieving functional recovery postinjury. Mil. Med. Res. 2020, 7, 30. [Google Scholar] [CrossRef]
- Mar, F.M.; Simoes, A.R.; Rodrigo, I.S.; Sousa, M.M. Inhibitory Injury Signaling Represses Axon Regeneration after Dorsal Root Injury. Mol. Neurobiol. 2016, 53, 4596–4605. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, B.; Eva, R. Promoting axon regeneration in the central nervous system by increasing PI3-kinase signaling. Neural Regen. Res. 2022, 17, 1172–1182. [Google Scholar] [CrossRef]
- Huebner, E.A.; Strittmatter, S.M. Axon regeneration in the peripheral and central nervous systems. Results Probl. Cell Differ. 2009, 48, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.Y.; Snider, W.D. Different signaling pathways mediate regenerative versus developmental sensory axon growth. J. Neurosci. 2001, 21, RC164. [Google Scholar] [CrossRef]
- Hausott, B.; Klimaschewski, L. Promotion of Peripheral Nerve Regeneration by Stimulation of the Extracellular Signal-Regulated Kinase (ERK) Pathway. Anat. Rec. 2019, 302, 1261–1267. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Roux, P.P.; Barker, P.A. Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef]
- Gutierrez, H.; Davies, A.M. Regulation of neural process growth, elaboration and structural plasticity by NF-kappa B. Trends Neurosci. 2011, 34, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imielski, Y.; Schwamborn, J.C.; Luningschror, P.; Heimann, P.; Holzberg, M.; Werner, H.; Leske, O.; Puschel, A.W.; Memet, S.; Heumann, R.; et al. Regrowing the adult brain: NF-kappaB controls functional circuit formation and tissue homeostasis in the dentate gyrus. PLoS ONE 2012, 7, e30838. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-kappaB Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Weih, F.; Haenold, R. Role of nuclear factor kappa B in central nervous system regeneration. Neural Regen. Res. 2014, 9, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Klimaschewski, L.; Hausott, B.; Angelov, D.N. The pros and cons of growth factors and cytokines in peripheral axon regeneration. Int. Rev. Neurobiol. 2013, 108, 137–171. [Google Scholar] [CrossRef]
- Klimaschewski, L.; Claus, P. Fibroblast Growth Factor Signalling in the Diseased Nervous System. Mol. Neurobiol. 2021, 58, 3884–3902. [Google Scholar] [CrossRef]
- Romano, R.; Bucci, C. Role of EGFR in the Nervous System. Cells 2020, 9, 1887. [Google Scholar] [CrossRef]
- Traverse, S.; Gomez, N.; Paterson, H.; Marshall, C.; Cohen, P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem. J. 1992, 288, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Hausott, B.; Schlick, B.; Vallant, N.; Dorn, R.; Klimaschewski, L. Promotion of neurite outgrowth by fibroblast growth factor receptor 1 overexpression and lysosomal inhibition of receptor degradation in pheochromocytoma cells and adult sensory neurons. Neuroscience 2008, 153, 461–473. [Google Scholar] [CrossRef]
- Dailey, L.; Ambrosetti, D.; Mansukhani, A.; Basilico, C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005, 16, 233–247. [Google Scholar] [CrossRef]
- Hausott, B.; Vallant, N.; Auer, M.; Yang, L.; Dai, F.; Brand-Saberi, B.; Klimaschewski, L. Sprouty2 down-regulation promotes axon growth by adult sensory neurons. Mol. Cell. Neurosci. 2009, 42, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Zhong, J.; Snider, W.D. Raf and akt mediate distinct aspects of sensory axon growth. Neuron 2002, 35, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Atwal, J.K.; Massie, B.; Miller, F.D.; Kaplan, D.R. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and PI3-kinase. Neuron 2000, 27, 265–277. [Google Scholar] [CrossRef]
- Atwal, J.K.; Singh, K.K.; Tessier-Lavigne, M.; Miller, F.D.; Kaplan, D.R. Semaphorin 3F antagonizes neurotrophin-induced phosphatidylinositol 3-kinase and mitogen-activated protein kinase kinase signaling: A mechanism for growth cone collapse. J. Neurosci. 2003, 23, 7602–7609. [Google Scholar] [CrossRef] [Green Version]
- Mograbi, B.; Bocciardi, R.; Bourget, I.; Busca, R.; Rochet, N.; Farahi-Far, D.; Juhel, T.; Rossi, B. Glial cell line-derived neurotrophic factor-stimulated phosphatidylinositol 3-kinase and Akt activities exert opposing effects on the ERK pathway: Importance for the rescue of neuroectodermic cells. J. Biol. Chem. 2001, 276, 45307–45319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolcet, X.; Soler, R.M.; Gould, T.W.; Egea, J.; Oppenheim, R.W.; Comella, J.X. Cytokines promote motoneuron survival through the janus kinase-dependent activation of the phosphatidylinositol 3-kinase pathway. Mol. Cell. Neurosci. 2001, 18, 619–631. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Hausott, B.; Kurnaz, I.; Gajovic, S.; Klimaschewski, L. Signaling by neuronal tyrosine kinase receptors: Relevance for development and regeneration. Anat. Rec. 2009, 292, 1976–1985. [Google Scholar] [CrossRef]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta 2007, 1773, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Sjogreen, B.; Wiklund, P.; Ekstrom, P.A. Mitogen activated protein kinase inhibition by PD98059 blocks nerve growth factor stimulated axonal outgrowth from adult mouse dorsal root ganglia in vitro. Neuroscience 2000, 100, 407–416. [Google Scholar] [CrossRef]
- Tucker, B.A.; Rahimtula, M.; Mearow, K.M. Src and FAK are key early signalling intermediates required for neurite growth in NGF-responsive adult DRG neurons. Cell Signal. 2008, 20, 241–257. [Google Scholar] [CrossRef]
- Chierzi, S.; Ratto, G.M.; Verma, P.; Fawcett, J.W. The ability of axons to regenerate their growth cones depends on axonal type and age, and is regulated by calcium, cAMP and ERK. Eur. J. Neurosci. 2005, 21, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.G.; Gordon-Weeks, P.R. The MAP kinase pathway is upstream of the activation of GSK3beta that enables it to phosphorylate MAP1B and contributes to the stimulation of axon growth. Mol. Cell Neurosci. 2005, 28, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Agthong, S.; Kaewsema, A.; Tanomsridejchai, N.; Chentanez, V. Activation of MAPK ERK in peripheral nerve after injury. BMC Neurosci. 2006, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agthong, S.; Koonam, J.; Kaewsema, A.; Chentanez, V. Inhibition of MAPK ERK impairs axonal regeneration without an effect on neuronal loss after nerve injury. Neurol. Res. 2009, 31, 1068–1074. [Google Scholar] [CrossRef]
- Huang, H.T.; Sun, Z.G.; Liu, H.W.; Ma, J.T.; Hu, M. ERK/MAPK and PI3K/AKT signal channels simultaneously activated in nerve cell and axon after facial nerve injury. Saudi J. Biol. Sci. 2017, 24, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Kanning, K.; Cavanaugh, J.E.; Xia, Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J. Biol. Chem. 1999, 274, 22569–22580. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.N.; Tolkovsky, A.M. A role for MAPK/ERK in sympathetic neuron survival: Protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 1999, 19, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Jethwa, N.; Chung, G.H.; Lete, M.G.; Alonso, A.; Byrne, R.D.; Calleja, V.; Larijani, B. Endomembrane PtdIns(3,4,5)P3 activates the PI3K-Akt pathway. J. Cell Sci. 2015, 128, 3456–3465. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Hur, E.M.; Zhou, F.Q. Coordinating Gene Expression and Axon Assembly to Control Axon Growth: Potential Role of GSK3 Signaling. Front. Mol. Neurosci. 2012, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.M.; Tucker, B.A.; Rahimtula, M.; Mearow, K.M. The synergistic effects of NGF and IGF-1 on neurite growth in adult sensory neurons: Convergence on the PI 3-kinase signaling pathway. J. Neurochem. 2003, 86, 1116–1128. [Google Scholar] [CrossRef]
- Kimpinski, K.; Mearow, K. Neurite growth promotion by nerve growth factor and insulin-like growth factor-1 in cultured adult sensory neurons: Role of phosphoinositide 3-kinase and mitogen activated protein kinase. J. Neurosci. Res. 2001, 63, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Edstrom, A.; Ekstrom, P.A. Role of phosphatidylinositol 3-kinase in neuronal survival and axonal outgrowth of adult mouse dorsal root ganglia explants. J. Neurosci. Res. 2003, 74, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, B.; Barber, A.C.; Evans, R.S.; Pearson, C.S.; Fuchs, J.; MacQueen, A.R.; van Erp, S.; Haenzi, B.; Hulshof, L.A.; Osborne, A.; et al. PI 3-kinase delta enhances axonal PIP3 to support axon regeneration in the adult CNS. EMBO Mol. Med. 2020, 12, e11674. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Sabit, H.; Oya, T.; Ishii, Y.; Hamashima, T.; Tokunaga, A.; Ishizawa, S.; Jie, S.; Kurashige, Y.; Matsushima, T.; et al. Activation of MAP kinases, Akt and PDGF receptors in injured peripheral nerves. J. Peripher. Nerv. Syst. 2009, 14, 165–176. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef]
- Crowder, R.J.; Freeman, R.S. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 1998, 18, 2933–2943. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Fan, H.; Li, F.; Skeeters, S.S.; Krishnamurthy, V.V.; Song, Y.; Zhang, K. Optical control of ERK and AKT signaling promotes axon regeneration and functional recovery of PNS and CNS in Drosophila. Elife 2020, 9, e57395. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamsuwan, S.; Klimaschewski, L.; Hausott, B. Simultaneous Knockdown of Sprouty2 and PTEN Promotes Axon Elongation of Adult Sensory Neurons. Front. Cell Neurosci. 2019, 13, 583. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Du, T.; Li, B.M.; Rong, Y.; Verkhratsky, A.; Peng, L. Crosstalk Between MAPK/ERK and PI3K/AKT Signal Pathways During Brain Ischemia/Reperfusion. ASN Neuro 2015, 7, 1759091415602463. [Google Scholar] [CrossRef]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Hacohen, N.; Kramer, S.; Sutherland, D.; Hiromi, Y.; Krasnow, M.A. sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell 1998, 92, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Minowada, G.; Jarvis, L.A.; Chi, C.L.; Neubuser, A.; Sun, X.; Hacohen, N.; Krasnow, M.A.; Martin, G.R. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 1999, 126, 4465–4475. [Google Scholar] [CrossRef]
- Tefft, D.; Lee, M.; Smith, S.; Crowe, D.L.; Bellusci, S.; Warburton, D. mSprouty2 inhibits FGF10-activated MAP kinase by differentially binding to upstream target proteins. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L700–706. [Google Scholar] [CrossRef] [Green Version]
- de Maximy, A.A.; Nakatake, Y.; Moncada, S.; Itoh, N.; Thiery, J.P.; Bellusci, S. Cloning and expression pattern of a mouse homologue of drosophila sprouty in the mouse embryo. Mech. Dev. 1999, 81, 213–216. [Google Scholar] [CrossRef]
- Leeksma, O.C.; Van Achterberg, T.A.; Tsumura, Y.; Toshima, J.; Eldering, E.; Kroes, W.G.; Mellink, C.; Spaargaren, M.; Mizuno, K.; Pannekoek, H.; et al. Human sprouty 4, a new ras antagonist on 5q31, interacts with the dual specificity kinase TESK1. Eur. J. Biochem. 2002, 269, 2546–2556. [Google Scholar] [CrossRef]
- Gross, I.; Bassit, B.; Benezra, M.; Licht, J.D. Mammalian sprouty proteins inhibit cell growth and differentiation by preventing ras activation. J. Biol. Chem. 2001, 276, 46460–46468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.S.; Fong, C.W.; Lim, J.; Yusoff, P.; Low, B.C.; Langdon, W.Y.; Guy, G.R. Sprouty2 attenuates epidermal growth factor receptor ubiquitylation and endocytosis, and consequently enhances Ras/ERK signalling. EMBO J. 2002, 21, 4796–4808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, I.; Armant, O.; Benosman, S.; de Aguilar, J.L.; Freund, J.N.; Kedinger, M.; Licht, J.D.; Gaiddon, C.; Loeffler, J.P. Sprouty2 inhibits BDNF-induced signaling and modulates neuronal differentiation and survival. Cell Death Differ. 2007, 14, 1802–1812. [Google Scholar] [CrossRef]
- Taketomi, T.; Yoshiga, D.; Taniguchi, K.; Kobayashi, T.; Nonami, A.; Kato, R.; Sasaki, M.; Sasaki, A.; Ishibashi, H.; Moriyama, M.; et al. Loss of mammalian Sprouty2 leads to enteric neuronal hyperplasia and esophageal achalasia. Nat. Neurosci. 2005, 8, 855–857. [Google Scholar] [CrossRef]
- Impagnatiello, M.A.; Weitzer, S.; Gannon, G.; Compagni, A.; Cotten, M.; Christofori, G. Mammalian sprouty-1 and -2 are membrane-anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J. Cell Biol. 2001, 152, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, J.E.; Hall, A.B.; Yatsula, B.A.; Bar-Sagi, D. The bimodal regulation of epidermal growth factor signaling by human Sprouty proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 6041–6046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusoff, P.; Lao, D.H.; Ong, S.H.; Wong, E.S.; Lim, J.; Lo, T.L.; Leong, H.F.; Fong, C.W.; Guy, G.R. Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. J. Biol. Chem. 2002, 277, 3195–3201. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Nishida, E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 2002, 4, 850–858. [Google Scholar] [CrossRef]
- Rathmanner, N.; Haigl, B.; Vanas, V.; Doriguzzi, A.; Gsur, A.; Sutterluty-Fall, H. Sprouty2 but not Sprouty4 is a potent inhibitor of cell proliferation and migration of osteosarcoma cells. FEBS Lett. 2013, 587, 2597–2605. [Google Scholar] [CrossRef] [Green Version]
- Edwin, F.; Singh, R.; Endersby, R.; Baker, S.J.; Patel, T.B. The tumor suppressor PTEN is necessary for human Sprouty 2-mediated inhibition of cell proliferation. J. Biol. Chem. 2006, 281, 4816–4822. [Google Scholar] [CrossRef]
- Patel, R.; Gao, M.; Ahmad, I.; Fleming, J.; Singh, L.B.; Rai, T.S.; McKie, A.B.; Seywright, M.; Barnetson, R.J.; Edwards, J.; et al. Sprouty2, PTEN, and PP2A interact to regulate prostate cancer progression. J. Clin. Investig. 2013, 123, 1157–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poppleton, H.M.; Edwin, F.; Jaggar, L.; Ray, R.; Johnson, L.R.; Patel, T.B. Sprouty regulates cell migration by inhibiting the activation of Rac1 GTPase. Biochem. Biophys. Res. Commun. 2004, 323, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, S.; Reddi, A.L.; Aggarwal, P.; Ambardekar, C.; Canciani, B.; Kim, M.K.; Hix, L.; Vilimas, T.; Mason, J.; Basson, M.A.; et al. Sprouty proteins inhibit receptor-mediated activation of phosphatidylinositol-specific phospholipase C. Mol. Biol. Cell 2010, 21, 3487–3496. [Google Scholar] [CrossRef] [Green Version]
- Marvaldi, L.; Thongrong, S.; Kozlowska, A.; Irschick, R.; Pritz, C.O.; Baumer, B.; Ronchi, G.; Geuna, S.; Hausott, B.; Klimaschewski, L. Enhanced axon outgrowth and improved long-distance axon regeneration in sprouty2 deficient mice. Dev. Neurobiol. 2015, 75, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Miyazaki, S.; Tanimura, S.; Kohno, M. Efficient suppression of FGF-2-induced ERK activation by the cooperative interaction among mammalian Sprouty isoforms. J. Cell Sci. 2005, 118, 5861–5871. [Google Scholar] [CrossRef] [Green Version]
- Hausott, B.; Vallant, N.; Schlick, B.; Auer, M.; Nimmervoll, B.; Obermair, G.J.; Schwarzer, C.; Dai, F.; Brand-Saberi, B.; Klimaschewski, L. Sprouty2 and -4 regulate axon outgrowth by hippocampal neurons. Hippocampus 2012, 22, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Edwin, F.; Anderson, K.; Ying, C.; Patel, T.B. Intermolecular interactions of Sprouty proteins and their implications in development and disease. Mol. Pharmacol. 2009, 76, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Masoumi-Moghaddam, S.; Amini, A.; Morris, D.L. The developing story of Sprouty and cancer. Cancer Metastasis Rev. 2014, 33, 695–720. [Google Scholar] [CrossRef] [Green Version]
- Lao, D.H.; Yusoff, P.; Chandramouli, S.; Philp, R.J.; Fong, C.W.; Jackson, R.A.; Saw, T.Y.; Yu, C.Y.; Guy, G.R. Direct binding of PP2A to Sprouty2 and phosphorylation changes are a prerequisite for ERK inhibition downstream of fibroblast growth factor receptor stimulation. J. Biol. Chem. 2007, 282, 9117–9126. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Matsumoto, K.; Nishida, E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J. Biol. Chem. 2004, 279, 22992–22995. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.S.; Lim, J.; Low, B.C.; Chen, Q.; Guy, G.R. Evidence for direct interaction between Sprouty and Cbl. J. Biol. Chem. 2001, 276, 5866–5875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeau, R.J.; Toher, J.L.; Yang, X.; Kovalenko, D.; Friesel, R. Regulation of Sprouty2 stability by mammalian Seven-in-Absentia homolog 2. J. Cell Biochem. 2007, 100, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Rane, S.; Lypowy, J.; He, M.; Chen, I.Y.; Vashistha, H.; Yan, L.; Malhotra, A.; Vatner, D.; Abdellatif, M. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol. Biol. Cell 2008, 19, 3272–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, H.J.; Kim, Y.J.; Chun, K.R.; Woo, Y.M.; Park, S.J.; Jeong, J.A.; Jo, S.H.; Kim, T.H.; Min, H.S.; Chae, J.S.; et al. Downregulation of Spry2 by miR-21 triggers malignancy in human gliomas. Oncogene 2011, 30, 2433–2442. [Google Scholar] [CrossRef] [Green Version]
- Suzuki-Hirano, A.; Sato, T.; Nakamura, H. Regulation of isthmic Fgf8 signal by sprouty2. Development 2005, 132, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Yaguchi, Y.; Echevarria, D.; Martinez, S.; Basson, M.A. Sprouty genes prevent excessive FGF signalling in multiple cell types throughout development of the cerebellum. Development 2011, 138, 2957–2968. [Google Scholar] [CrossRef] [Green Version]
- Shim, K.; Minowada, G.; Coling, D.E.; Martin, G.R. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev. Cell 2005, 8, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Ciganovic, N.; Warren, R.L.; Keceli, B.; Jacob, S.; Fridberger, A.; Reichenbach, T. Static length changes of cochlear outer hair cells can tune low-frequency hearing. PLoS Comput. Biol. 2018, 14, e1005936. [Google Scholar] [CrossRef] [Green Version]
- Jacques, B.E.; Montcouquiol, M.E.; Layman, E.M.; Lewandoski, M.; Kelley, M.W. Fgf8 induces pillar cell fate and regulates cellular patterning in the mammalian cochlea. Development 2007, 134, 3021–3029. [Google Scholar] [CrossRef] [Green Version]
- Strickland, I.T.; Richards, L.; Holmes, F.E.; Wynick, D.; Uney, J.B.; Wong, L.F. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS ONE 2011, 6, e23423. [Google Scholar] [CrossRef] [Green Version]
- Thongrong, S.; Hausott, B.; Marvaldi, L.; Agostinho, A.S.; Zangrandi, L.; Burtscher, J.; Fogli, B.; Schwarzer, C.; Klimaschewski, L. Sprouty2 and -4 hypomorphism promotes neuronal survival and astrocytosis in a mouse model of kainic acid induced neuronal damage. Hippocampus 2016, 26, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Klimaschewski, L.; Sueiro, B.P.; Millan, L.M. siRNA mediated down-regulation of Sprouty2/4 diminishes ischemic brain injury. Neurosci. Lett. 2016, 612, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musatov, S.; Roberts, J.; Brooks, A.I.; Pena, J.; Betchen, S.; Pfaff, D.W.; Kaplitt, M.G. Inhibition of neuronal phenotype by PTEN in PC12 cells. Proc. Natl. Acad. Sci. USA 2004, 101, 3627–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB Receptor Signaling Underlies Corticostriatal Dysfunction in Huntington’s Disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Dressler, G.R. PTEN modulates GDNF/RET mediated chemotaxis and branching morphogenesis in the developing kidney. Dev. Biol. 2007, 307, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Guntur, A.R.; Reinhold, M.I.; Cuellar, J., Jr.; Naski, M.C. Conditional ablation of Pten in osteoprogenitors stimulates FGF signaling. Development 2011, 138, 1433–1444. [Google Scholar] [CrossRef] [Green Version]
- Hertzler-Schaefer, K.; Mathew, G.; Somani, A.K.; Tholpady, S.; Kadakia, M.P.; Chen, Y.; Spandau, D.F.; Zhang, X. Pten loss induces autocrine FGF signaling to promote skin tumorigenesis. Cell Rep. 2014, 6, 818–826. [Google Scholar] [CrossRef] [Green Version]
- Vivanco, I.; Rohle, D.; Versele, M.; Iwanami, A.; Kuga, D.; Oldrini, B.; Tanaka, K.; Dang, J.; Kubek, S.; Palaskas, N.; et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc. Natl. Acad. Sci. USA 2010, 107, 6459–6464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diekmann, H.; Kalbhen, P.; Fischer, D. Active mechanistic target of rapamycin plays an ancillary rather than essential role in zebrafish CNS axon regeneration. Front. Cell. Neurosci. 2015, 9, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Shen, D.; Wang, Y.; Gong, L.; Tang, X.; Yu, B.; Gu, X.; Ding, F. microRNA-222 targeting PTEN promotes neurite outgrowth from adult dorsal root ganglion neurons following sciatic nerve transection. PLoS ONE 2012, 7, e44768. [Google Scholar] [CrossRef]
- Li, B.; Sun, H. MiR-26a promotes neurite outgrowth by repressing PTEN expression. Mol. Med. Rep. 2013, 8, 676–680. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Chen, F.; Ge, X.; Tan, J.; Lei, P.; Zhang, J. miR-21 alleviated apoptosis of cortical neurons through promoting PTEN-Akt signaling pathway in vitro after experimental traumatic brain injury. Brain Res. 2014, 1582, 12–20. [Google Scholar] [CrossRef]
- Wang, W.M.; Lu, G.; Su, X.W.; Lyu, H.; Poon, W.S. MicroRNA-182 Regulates Neurite Outgrowth Involving the PTEN/AKT Pathway. Front. Cell Neurosci. 2017, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Kar, A.N.; Lee, S.J.; Sahoo, P.K.; Thames, E.; Yoo, S.; Houle, J.D.; Twiss, J.L. MicroRNAs 21 and 199a-3p Regulate Axon Growth Potential through Modulation of Pten and mTor mRNAs. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, Y.X.; Zeng, Y.Q.; Zhang, S.F.; Teng, Z.Q.; Gao, J.; Saijilafu; Liu, C.M. miR-26a promotes axon regeneration in the mammalian central nervous system by suppressing PTEN expression. Acta Biochim. Biophys. Sin. 2021, 53, 758–765. [Google Scholar] [CrossRef]
- Van Themsche, C.; Leblanc, V.; Parent, S.; Asselin, E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J. Biol. Chem. 2009, 284, 20462–20466. [Google Scholar] [CrossRef] [Green Version]
- Christie, K.J.; Martinez, J.A.; Zochodne, D.W. Disruption of E3 ligase NEDD4 in peripheral neurons interrupts axon outgrowth: Linkage to PTEN. Mol. Cell Neurosci. 2012, 50, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Unsain, N.; Higgins, J.M.; Parker, K.N.; Johnstone, A.D.; Barker, P.A. XIAP regulates caspase activity in degenerating axons. Cell Rep. 2013, 4, 751–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drinjakovic, J.; Jung, H.; Campbell, D.S.; Strochlic, L.; Dwivedy, A.; Holt, C.E. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 2010, 65, 341–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F.; Sellers, W.R. The PTEN tumor suppressor protein: An antagonist of phosphoinositide 3-kinase signaling. Biochim. Biophys. Acta 2000, 1470, M21–35. [Google Scholar] [CrossRef]
- van Diepen, M.T.; Parsons, M.; Downes, C.P.; Leslie, N.R.; Hindges, R.; Eickholt, B.J. MyosinV controls PTEN function and neuronal cell size. Nat. Cell Biol. 2009, 11, 1191–1196. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.H.; Zhu, X.; Zhang, J.; Baker, S.J. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 12923–12928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Planchon, S.M.; Waite, K.A.; Eng, C. The nuclear affairs of PTEN. J. Cell Sci. 2008, 121, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Leclere, P.G.; Norman, E.; Groutsi, F.; Coffin, R.; Mayer, U.; Pizzey, J.; Tonge, D. Impaired axonal regeneration by isolectin B4-binding dorsal root ganglion neurons in vitro. J. Neurosci. 2007, 27, 1190–1199. [Google Scholar] [CrossRef]
- Christie, K.J.; Webber, C.A.; Martinez, J.A.; Singh, B.; Zochodne, D.W. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J. Neurosci. 2010, 30, 9306–9315. [Google Scholar] [CrossRef]
- Lachyankar, M.B.; Sultana, N.; Schonhoff, C.M.; Mitra, P.; Poluha, W.; Lambert, S.; Quesenberry, P.J.; Litofsky, N.S.; Recht, L.D.; Nabi, R.; et al. A role for nuclear PTEN in neuronal differentiation. J. Neurosci. 2000, 20, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Brosig, A.; Fuchs, J.; Ipek, F.; Kroon, C.; Schrotter, S.; Vadhvani, M.; Polyzou, A.; Ledderose, J.; van Diepen, M.; Holzhutter, H.G.; et al. The Axonal Membrane Protein PRG2 Inhibits PTEN and Directs Growth to Branches. Cell Rep. 2019, 29, 2028–2040e8. [Google Scholar] [CrossRef] [Green Version]
- Gutilla, E.A.; Steward, O. Selective neuronal PTEN deletion: Can we take the brakes off of growth without losing control? Neural Regen. Res. 2016, 11, 1201–1203. [Google Scholar] [CrossRef]
- Kath, C.; Goni-Oliver, P.; Muller, R.; Schultz, C.; Haucke, V.; Eickholt, B.; Schmoranzer, J. PTEN suppresses axon outgrowth by down-regulating the level of detyrosinated microtubules. PLoS ONE 2018, 13, e0193257. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.Y.; Sui, L.; Yamaguchi, F.; Kamitori, K.; Hirata, Y.; Suzuki, A.; Holley, M.; Tokuda, M. Role of phosphatase and tensin homolog in the development of the mammalian auditory system. Neuroreport 2010, 21, 731–735. [Google Scholar] [CrossRef]
- Dong, Y.; Sui, L.; Yamaguchi, F.; Kamitori, K.; Hirata, Y.; Hossain, M.A.; Suzuki, A.; Holley, M.C.; Tokuda, M. Phosphatase and Tensin Homolog Deleted on Chromosome 10 Regulates Sensory Cell Proliferation and Differentiation of Hair Bundles in the Mammalian Cochlea. Neuroscience 2010, 170, 1304–1313. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, J.; Jin, Y.C.; Hou, C.Z.; Zong, W.; Lu, T.T.; Li, H.S.; Gao, J.G. PTEN regulation of the proliferation and differentiation of auditory progenitors through the PTEN/PI3K/Akt-signaling pathway in mice. Neuroreport 2014, 25, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Yang, L.M.; Ornitz, D.M. FGF20-FGFR1 signaling through MAPK and PI3K controls sensory progenitor differentiation in the organ of Corti. Dev. Dyn. 2021, 250, 134–144. [Google Scholar] [CrossRef]
- Huh, S.H.; Jones, J.; Warchol, M.E.; Ornitz, D.M. Differentiation of the Lateral Compartment of the Cochlea Requires a Temporally Restricted FGF20 Signal. PLoS Biol. 2012, 10, e1001231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Ray, C.A.; Bermingham-McDonogh, O. Fgf20 is required for sensory epithelial specification in the developing cochlea. J. Neurosci. 2008, 28, 5991–5999. [Google Scholar] [CrossRef] [PubMed]
- Chadborn, N.H.; Ahmed, A.I.; Holt, M.R.; Prinjha, R.; Dunn, G.A.; Jones, G.E.; Eickholt, B.J. PTEN couples Sema3A signalling to growth cone collapse. J. Cell Sci. 2006, 119, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Xiang, J.; Wu, J.; He, B.; Lin, T.; Zhu, Q.; Liu, X.; Zheng, C. Expression patterns and role of PTEN in rat peripheral nerve development and injury. Neurosci. Lett. 2018, 676, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 2010, 13, 1075–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zukor, K.; Belin, S.; Wang, C.; Keelan, N.; Wang, X.; He, Z. Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J. Neurosci. 2013, 33, 15350–15361. [Google Scholar] [CrossRef]
- Lewandowski, G.; Steward, O. AAVshRNA-mediated suppression of PTEN in adult rats in combination with salmon fibrin administration enables regenerative growth of corticospinal axons and enhances recovery of voluntary motor function after cervical spinal cord injury. J. Neurosci. 2014, 34, 9951–9962. [Google Scholar] [CrossRef] [Green Version]
- Danilov, C.A.; Steward, O. Conditional genetic deletion of PTEN after a spinal cord injury enhances regenerative growth of CST axons and motor function recovery in mice. Exp. Neurol. 2015, 266, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Zheng, S.; Zhang, Q.; Li, S.; Gao, X.; Wang, J.; Jiang, L.; Liu, K. Pten Deletion Promotes Regrowth of Corticospinal Tract Axons 1 Year after Spinal Cord Injury. J. Neurosci. 2015, 35, 9754–9763. [Google Scholar] [CrossRef] [Green Version]
- Gutilla, E.A.; Buyukozturk, M.M.; Steward, O. Long-term consequences of conditional genetic deletion of PTEN in the sensorimotor cortex of neonatal mice. Exp. Neurol. 2016, 279, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 307–319. [Google Scholar] [CrossRef]
- Singh, B.; Singh, V.; Krishnan, A.; Koshy, K.; Martinez, J.A.; Cheng, C.; Almquist, C.; Zochodne, D.W. Regeneration of diabetic axons is enhanced by selective knockdown of the PTEN gene. Brain 2014, 137, 1051–1067. [Google Scholar] [CrossRef] [Green Version]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.C.; Byrne, R.D.; Vilar, R.; Woscholski, R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett. 2004, 566, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Abak, A.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Sayad, A.; Taheri, M. Regulatory role of microRNAs on PTEN signaling. Biomed. Pharmacother. 2021, 133, 110986. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, Z.; Zhang, J. Transcription Factor E2F1 Aggravates Neurological Injury in Ischemic Stroke via microRNA-122-Targeted Sprouty2. Neuropsychiatr. Dis. Treat. 2020, 16, 2633–2647. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Xu, W.; Zhou, P.; Gao, P.; Jiang, S.; Lobie, P.E.; Zhu, T. c-MYC-regulated miR-23a/24-2/27a cluster promotes mammary carcinoma cell invasion and hepatic metastasis by targeting Sprouty2. J. Biol. Chem. 2013, 288, 18121–18133. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Yang, M.; Yang, H.; Chang, R.; Fang, F.; Yang, L. miR-330-5p targets SPRY2 to promote hepatocellular carcinoma progression via MAPK/ERK signaling. Oncogenesis 2018, 7, 90. [Google Scholar] [CrossRef]
- Liu, Y.; Li, S.; Liu, Y.; Lv, X.; Zhou, Q. MicroRNA-124 facilitates lens epithelial cell apoptosis by inhibiting SPRY2 and MMP-2. Mol. Med. Rep. 2021, 23, 381. [Google Scholar] [CrossRef]
- Gallaher, Z.R.; Steward, O. Modest enhancement of sensory axon regeneration in the sciatic nerve with conditional co-deletion of PTEN and SOCS3 in the dorsal root ganglia of adult mice. Exp. Neurol. 2018, 303, 120–133. [Google Scholar] [CrossRef]
- Sun, F.; Park, K.K.; Belin, S.; Wang, D.; Lu, T.; Chen, G.; Zhang, K.; Yeung, C.; Feng, G.; Yankner, B.A.; et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 2011, 480, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.D.; Sun, F.; Park, K.K.; Cai, B.; Wang, C.; Kuwako, K.; Martinez-Carrasco, I.; Connolly, L.; He, Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 2009, 64, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Yungher, B.J.; Luo, X.; Salgueiro, Y.; Blackmore, M.G.; Park, K.K. Viral vector-based improvement of optic nerve regeneration: Characterization of individual axons’ growth patterns and synaptogenesis in a visual target. Gene Ther. 2015, 22, 811–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lima, S.; Koriyama, Y.; Kurimoto, T.; Oliveira, J.T.; Yin, Y.; Li, Y.; Gilbert, H.Y.; Fagiolini, M.; Martinez, A.M.; Benowitz, L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc. Natl. Acad. Sci. USA 2012, 109, 9149–9154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibinger, M.; Zeitler, C.; Gobrecht, P.; Andreadaki, A.; Gisselmann, G.; Fischer, D. Transneuronal delivery of hyper-interleukin-6 enables functional recovery after severe spinal cord injury in mice. Nat. Commun. 2021, 12, 391. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hausott, B.; Glueckert, R.; Schrott-Fischer, A.; Klimaschewski, L. Signal Transduction Regulators in Axonal Regeneration. Cells 2022, 11, 1537. https://doi.org/10.3390/cells11091537
Hausott B, Glueckert R, Schrott-Fischer A, Klimaschewski L. Signal Transduction Regulators in Axonal Regeneration. Cells. 2022; 11(9):1537. https://doi.org/10.3390/cells11091537
Chicago/Turabian StyleHausott, Barbara, Rudolf Glueckert, Anneliese Schrott-Fischer, and Lars Klimaschewski. 2022. "Signal Transduction Regulators in Axonal Regeneration" Cells 11, no. 9: 1537. https://doi.org/10.3390/cells11091537
APA StyleHausott, B., Glueckert, R., Schrott-Fischer, A., & Klimaschewski, L. (2022). Signal Transduction Regulators in Axonal Regeneration. Cells, 11(9), 1537. https://doi.org/10.3390/cells11091537