
PTPRK, an EGFR Phosphatase, Is Decreased in CeD Biopsies and Intestinal Organoids

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Biopsies
2.2. Organoids
2.3. Immunofluorescence Staining of Biopsies
2.4. BrdU Cell Proliferation Assay
2.5. Western Blot
2.6. Crypt Epithelial Cell Proliferation Test
2.7. mRNA Analysis
2.8. PTPRK Silencing
2.9. PTPRK Overexpression
2.10. Statistical Analysis
3. Results
3.1. In Enterocytes from GCD–CeD and Pot–CeD Biopsies, PTPRK Is Decreased, pEGFR, pERK and Proliferation Are Increased
3.2. Intestinal Organoids from CeD Patients Reproduce the Increase inpEGFR, pERK and Proliferation Found in GCD–CeD and Pot–CeD Biopsies
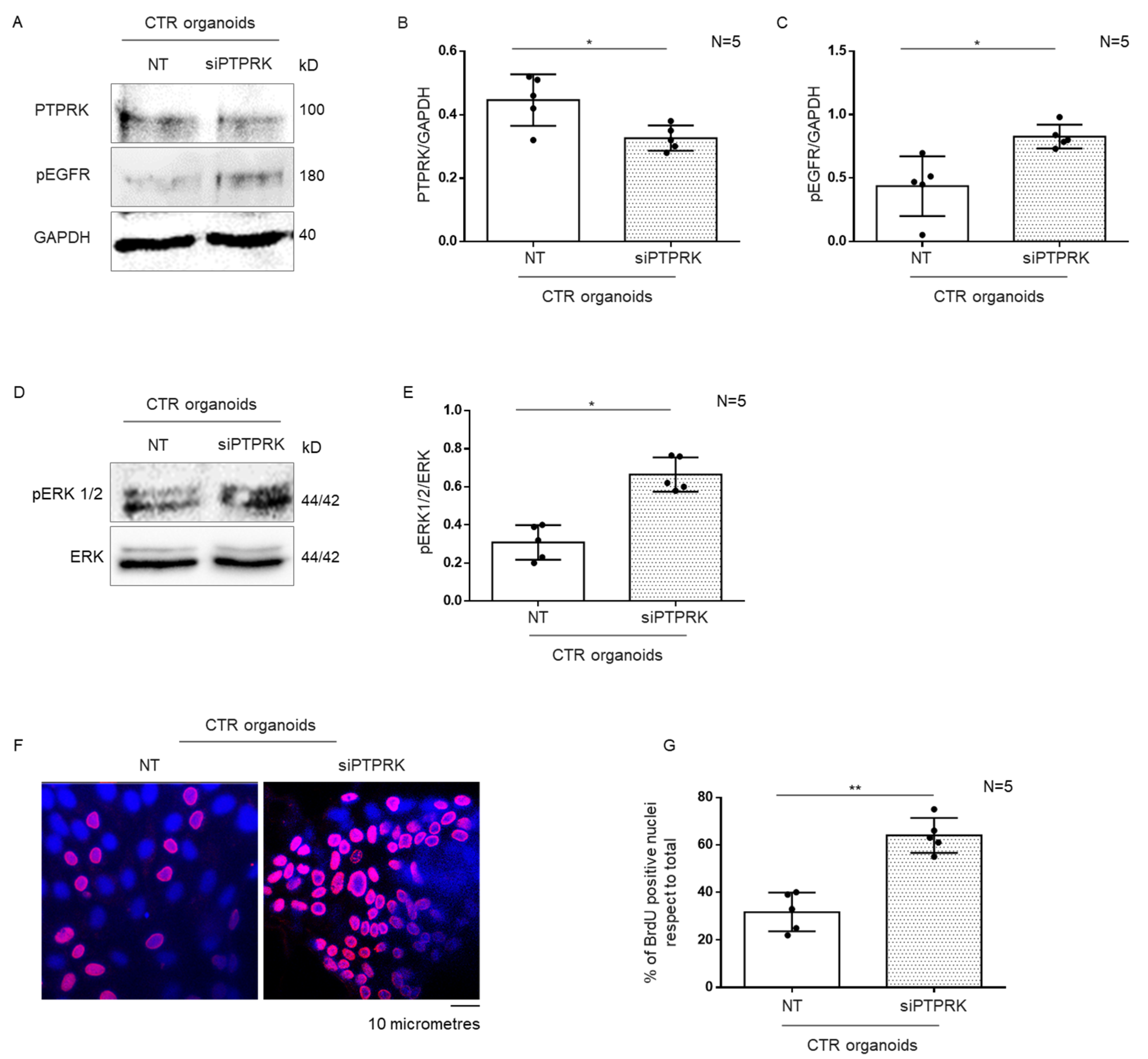
3.3. Silencing of PTPRK Protein in CTR Organoids Induced Increase in pEGFR, pERK and Proliferation
3.4. Overexpression of PTPRK Protein in CeD Organoids Reduced pEGFR, pERK and Proliferation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barone, M.V.; Auricchio, R.; Nanayakkara, M.; Greco, L.; Troncone, R.; Auricchio, S. Pivotal Role of Inflammation in Celiac Disease. Int. J. Mol. Sci. 2022, 23, 7177. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.N.; Loft, D.E.; Garner, V.; Gordon, D.B. Time/dose responses of coeliac mucosae to graded oral challenges with Frazer’s fraction III of gliadin. Eur. J. Gastroenterol. Hepatol. 1992, 4, 667–673. [Google Scholar]
- Mulder, C.J.; Mearin, M.L.; Peña, A.S. Clinical and pathological spectrum of coeliac disease. Gut 1993, 34, 1740–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, M.N.; Crowe, P.T. Morphology of the mucosal lesion in gluten sensitivity. Baillieres Clin. Gastroenterol. 1995, 9, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.V.; Troncone, R.; Auricchio, S. Gliadin peptides as triggers of the proliferative and stress/innate immune response of the celiac small intestinal mucosa. Int. J. Mol. Sci. 2014, 15, 20518–20537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juuti-Uusitalo, K.; Mäki, M.; Kainulainen, H.; Isola, J.; Kaukinen, K. Gluten affects epithelial differentiation-associated genes in small intestinal mucosa of coeliac patients. Clin. Exp. Immunol. 2007, 150, 294–305. [Google Scholar] [CrossRef]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Kosova, R.; Sarno, M.; Gaito, A.; Discepolo, V.; Troncone, R.; Auricchio, S.; Auricchio, R.; et al. Enterocyte proliferation and signaling are constitutively altered in celiac disease. PLoS ONE 2013, 8, e76006. [Google Scholar] [CrossRef] [Green Version]
- Lania, G.; Nanayakkara, M.; Maglio, M.; Auricchio, R.; Porpora, M.; Conte, M.; De Matteis, M.A.; Rizzo, R.; Luini, A.; Discepolo, V.; et al. Constitutive alterations in vesicular trafficking increase the sensitivity of cells from celiac disease patients to gliadin. Commun. Biol. 2019, 2, 190. [Google Scholar] [CrossRef] [Green Version]
- Chirdo, F.G.; Auricchio, S.; Troncone, R.; Barone, M.V. The gliadin p31-43 peptide: Inducer of multiple proinflammatory effects. Int. Rev. Cell Mol. Biol. 2021, 358, 165–205. [Google Scholar] [CrossRef]
- Barone, M.V.; Zanzi, D.; Maglio, M.; Nanayakkara, M.; Santagata, S.; Lania, G.; Miele, E.; Ribecco, M.T.; Maurano, F.; Auricchio, R.; et al. Gliadin-mediated proliferation and innate immune activation in celiac disease are due to alterations in vesicular trafficking. PLoS ONE 2011, 6, e17039. [Google Scholar] [CrossRef] [Green Version]
- Barone, M.V.; Nanayakkara, M.; Paolella, G.; Maglio, M.; Vitale, V.; Troiano, R.; Ribecco, M.T.; Lania, G.; Zanzi, D.; Santagata, S.; et al. Gliadin peptide P31-43 localises to endocytic vesicles and interferes with their maturation. PLoS ONE 2010, 5, e12246. [Google Scholar] [CrossRef] [PubMed]
- Taavela, J.; Viiri, K.; Popp, A.; Oittinen, M.; Dotsenko, V.; Peräaho, M.; Staff, S.; Sarin, J.; Leon, F.; Mäki, M.; et al. Histological, immunohistochemical and mRNA gene expression responses in coeliac disease patients challenged with gluten using PAXgene fixed paraffin-embedded duodenal biopsies. BMC Gastroenterol. 2019, 19, 189. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.V.; Gimigliano, A.; Castoria, G.; Paolella, G.; Maurano, F.; Paparo, F.; Maglio, M.; Mineo, A.; Miele, E.; Nanayakkara, M.; et al. Growth factor-like activity of gliadin, an alimentary protein: Implications for coeliac disease. Gut 2007, 56, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya, R.E.; Jury, J.; Bondar, C.; Verdu, E.F.; Chirdo, F.G. Intraluminal administration of poly I:C causes an enteropathy that is exacerbated by administration of oral dietary antigen. PLoS ONE 2014, 9, e99236. [Google Scholar] [CrossRef]
- Araya, R.E.; Gomez Castro, M.F.; Carasi, P.; McCarville, J.L.; Jury, J.; Mowat, A.M.; Verdu, E.F.; Chirdo, F.G. Mechanisms of innate immune activation by gluten peptide p31-43 in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G40–G49. [Google Scholar] [CrossRef] [Green Version]
- Abadie, V.; Sollid, L.M.; Barreiro, L.B.; Jabri, B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu. Rev. Immunol. 2011, 29, 493–525. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Gutierrez-Achury, J.; Kanduri, K.; Almeida, R.; Hrdlickova, B.; Zhernakova, D.V.; Westra, H.J.; Karjalainen, J.; Ricaño-Ponce, I.; Li, Y.; et al. Systematic annotation of celiac disease loci refines pathological pathways and suggests a genetic explanation for increased interferon-gamma levels. Hum. Mol. Genet 2015, 24, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Almeida, R.; Ricaño-Ponce, I.; Kumar, V.; Deelen, P.; Szperl, A.; Trynka, G.; Gutierrez-Achury, J.; Kanterakis, A.; Westra, H.J.; Franke, L.; et al. Fine mapping of the celiac disease-associated LPP locus reveals a potential functional variant. Hum. Mol. Genet 2014, 23, 2481–2489. [Google Scholar] [CrossRef] [Green Version]
- Nanayakkara, M.; Kosova, R.; Lania, G.; Sarno, M.; Gaito, A.; Galatola, M.; Greco, L.; Cuomo, M.; Troncone, R.; Auricchio, S.; et al. A celiac cellular phenotype, with altered LPP sub-cellular distribution, is inducible in controls by the toxic gliadin peptide P31-43. PLoS ONE 2013, 8, e79763. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.; Pasedag, S.M.; Butt, E. Cell Adhesion and Transcriptional Activity—Defining the Role of the Novel Protooncogene LPP. Transl. Oncol. 2009, 2, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Discepolo, V.; Lania, G.; Ten Eikelder, M.L.G.; Nanayakkara, M.; Sepe, L.; Tufano, R.; Troncone, R.; Auricchio, S.; Auricchio, R.; Paolella, G.; et al. Pediatric Celiac Disease Patients Show Alterations of Dendritic Cell Shape and Actin Rearrangement. Int. J. Mol. Sci. 2021, 22, 2708. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, G.W.; Young, K.A.; Edgar, J.R.; Antrobus, R.; Hay, I.M.; Liang, W.C.; Martinez-Martin, N.; Lin, W.; Deane, J.E.; Sharpe, H.J. The homophilic receptor PTPRK selectively dephosphorylates multiple junctional regulators to promote cell-cell adhesion. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Bondar, C.; Plaza-Izurieta, L.; Fernandez-Jimenez, N.; Irastorza, I.; Withoff, S.; Wijmenga, C.; Chirdo, F.; Bilbao, J.R. THEMIS and PTPRK in celiac intestinal mucosa: Coexpression in disease and after in vitro gliadin challenge. Eur. J. Hum. Genet 2014, 22, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonks, N.K.; Neel, B.G. From form to function: Signaling by protein tyrosine phosphatases. Cell 1996, 87, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.H. Cell signaling by protein tyrosine phosphorylation. Adv. Enzyme Regul. 1999, 39, 359–369. [Google Scholar] [CrossRef]
- Xu, Y.; Tan, L.J.; Grachtchouk, V.; Voorhees, J.J.; Fisher, G.J. Receptor-type protein-tyrosine phosphatase-kappa regulates epidermal growth factor receptor function. J. Biol. Chem. 2005, 280, 42694–42700. [Google Scholar] [CrossRef] [Green Version]
- Porpora, M.; Conte, M.; Lania, G.; Bellomo, C.; Rapacciuolo, L.; Chirdo, F.G.; Auricchio, R.; Troncone, R.; Auricchio, S.; Barone, M.V.; et al. Inflammation Is Present, Persistent and More Sensitive to Proinflammatory Triggers in Celiac Disease Enterocytes. Int. J. Mol. Sci. 2022, 23, 1973. [Google Scholar] [CrossRef]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Discepolo, V.; Sarno, M.; Gaito, A.; Troncone, R.; Auricchio, S.; Auricchio, R.; Barone, M.V. An undigested gliadin peptide activates innate immunity and proliferative signaling in enterocytes: The role in celiac disease. Am. J. Clin. Nutr. 2013, 98, 1123–1135. [Google Scholar] [CrossRef] [Green Version]
- Li, M.Y.; Lai, P.L.; Chou, Y.T.; Chi, A.P.; Mi, Y.Z.; Khoo, K.H.; Chang, G.D.; Wu, C.W.; Meng, T.C.; Chen, G.C. Protein tyrosine phosphatase PTPN3 inhibits lung cancer cell proliferation and migration by promoting EGFR endocytic degradation. Oncogene 2015, 34, 3791–3803. [Google Scholar] [CrossRef]
- Pike, K.A.; Tremblay, M.L. Protein Tyrosine Phosphatases: Regulators of CD4 T Cells in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2504. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xue, S.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Notch and TGF-β pathways cooperatively regulate receptor protein tyrosine phosphatase-κ (PTPRK) gene expression in human primary keratinocytes. Mol. Biol. Cell 2015, 26, 1199–1206. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Range Age (Years) | Sex | Biopsy (Marsh Classification *) | Serum AntiTG2 (U/mL) | Anti-Endomysial Antibody (EMA) |
|---|---|---|---|---|---|
| Controls (N = 20) | 9–17 | M = 8 F = 12 | 20 = T0 | 0–1.7 | Negative |
| GCD–CeD (N = 20) | 4–16 | M = 10 F = 10 | 6 = T3c 14 = T3 c/b | >50 | Positive |
| POT–CeD (N = 8) | 8–13 | M = 5 F = 3 | 6 = T0 2 = T1 | 9–60 | Positive |
| Patients | Range Age (Years) | Sex | Biopsy (Marsh Classification *) | Serum AntiTG2 (U/mL) | Anti-Endomysia Antibody (EMA) |
|---|---|---|---|---|---|
| Controls (N = 5) | 4–15 | M = 3 F = 2 | 5 = T0 | 0–1.9 | Negative |
| GCD–CeD (N = 5) | 8–17 | M = 2 F = 3 | 3 = T3c 2 = T3 c/b | >50 | Positive |
| Patients | Range Age (Years) | Sex | Biopsy (Marsh Classification *) | Ki67 % |
|---|---|---|---|---|
| Controls (N = 4) | 8–16 | M = 2 F = 2 | T0 | 22.5–33 |
| GCD–CeD (N = 4) | 3–15 | M = 3 F = 1 | T3c | 52.7–62.2 |
| POT–CeD (N = 4) | 2–14 | M = 1 F = 3 | T0/T1 | 37–53 |
| CeD–Patients | PTPRK | pEGFR | pERK | Ki67 Positive Nuclei |
|---|---|---|---|---|
| 12980 T3c | −2 | +7.8 | +2 | +2.2 |
| 12985 T3c | −4.6 | +9.3 | +3.6 | +2.3 |
| 12686 T3c | −2.3 | +6.3 | +3.5 | +1.9 |
| 12994 T3c | −2 | +7.1 | +2.6 | +2.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanayakkara, M.; Bellomo, C.; Furone, F.; Maglio, M.; Marano, A.; Lania, G.; Porpora, M.; Nicoletti, M.; Auricchio, S.; Barone, M.V. PTPRK, an EGFR Phosphatase, Is Decreased in CeD Biopsies and Intestinal Organoids. Cells 2023, 12, 115. https://doi.org/10.3390/cells12010115
Nanayakkara M, Bellomo C, Furone F, Maglio M, Marano A, Lania G, Porpora M, Nicoletti M, Auricchio S, Barone MV. PTPRK, an EGFR Phosphatase, Is Decreased in CeD Biopsies and Intestinal Organoids. Cells. 2023; 12(1):115. https://doi.org/10.3390/cells12010115
Chicago/Turabian StyleNanayakkara, Merlin, Claudia Bellomo, Francesca Furone, Mariantonia Maglio, Antonella Marano, Giuliana Lania, Monia Porpora, Martina Nicoletti, Salvatore Auricchio, and Maria Vittoria Barone. 2023. "PTPRK, an EGFR Phosphatase, Is Decreased in CeD Biopsies and Intestinal Organoids" Cells 12, no. 1: 115. https://doi.org/10.3390/cells12010115
APA StyleNanayakkara, M., Bellomo, C., Furone, F., Maglio, M., Marano, A., Lania, G., Porpora, M., Nicoletti, M., Auricchio, S., & Barone, M. V. (2023). PTPRK, an EGFR Phosphatase, Is Decreased in CeD Biopsies and Intestinal Organoids. Cells, 12(1), 115. https://doi.org/10.3390/cells12010115