
PAI-1 Overexpression in Valvular Interstitial Cells Contributes to Hypofibrinolysis in Aortic Stenosis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Laboratory Analysis
2.3. Aortic Valves Preparation
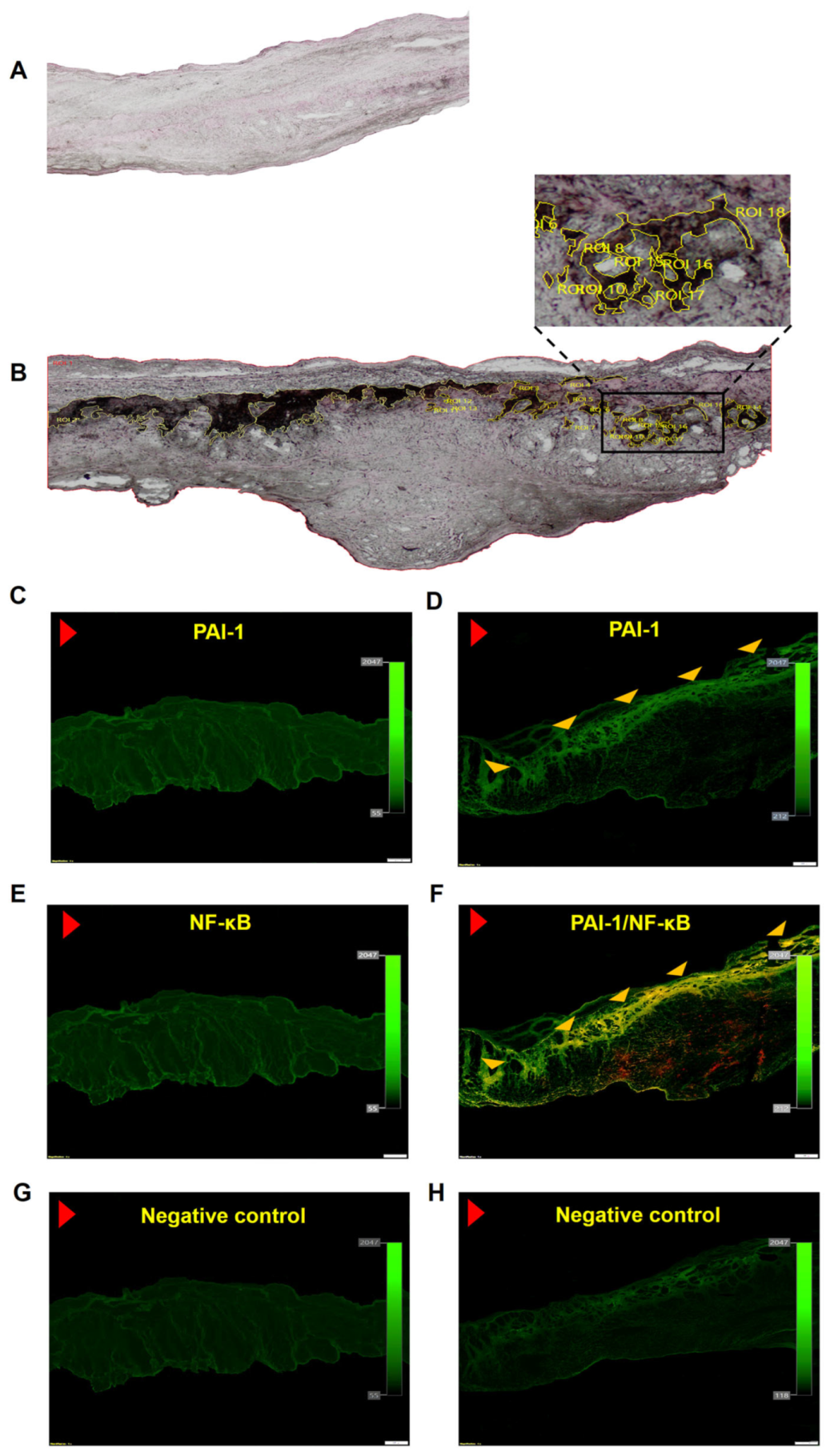
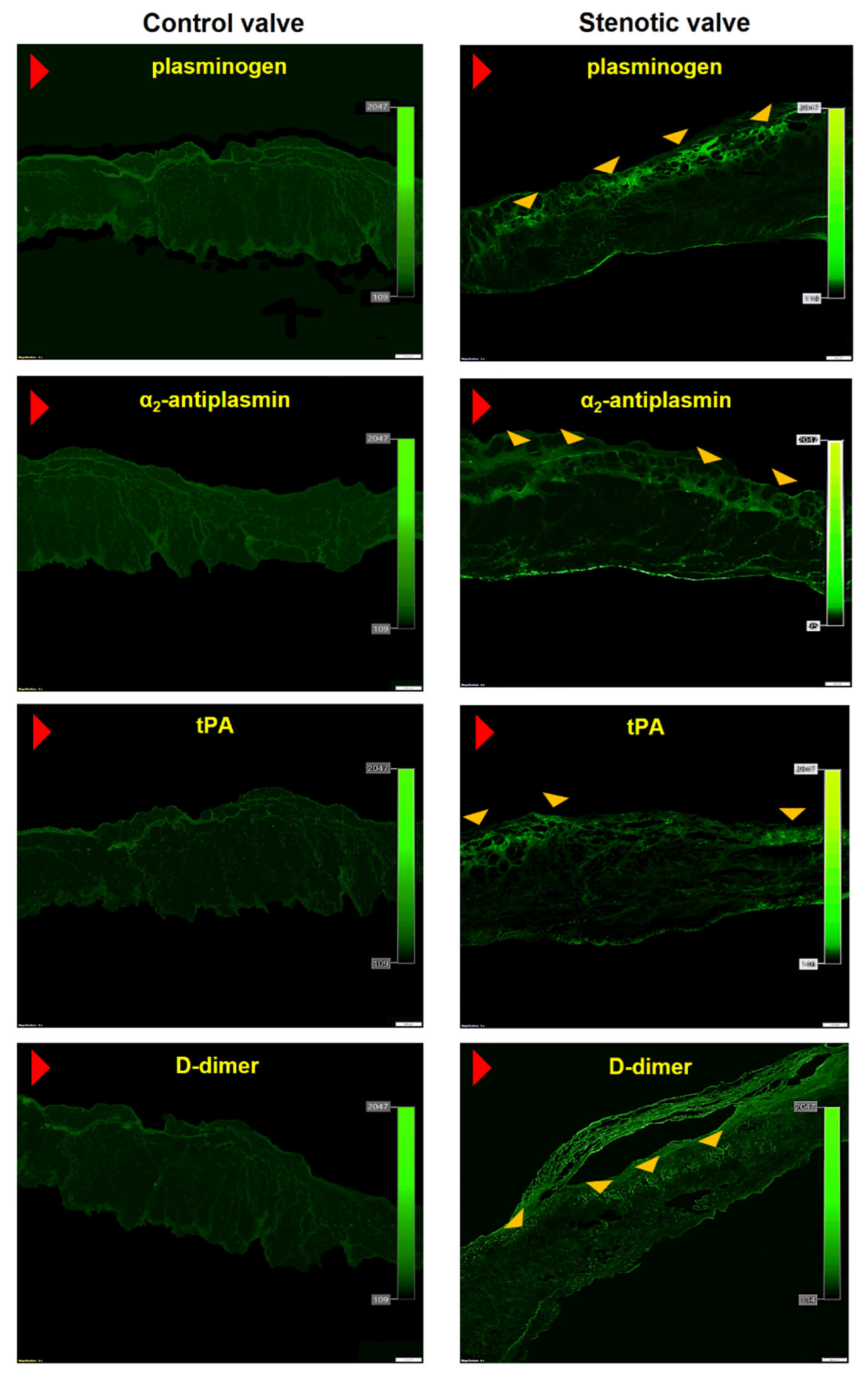
2.4. Histochemical and Immunofluorescence Staining
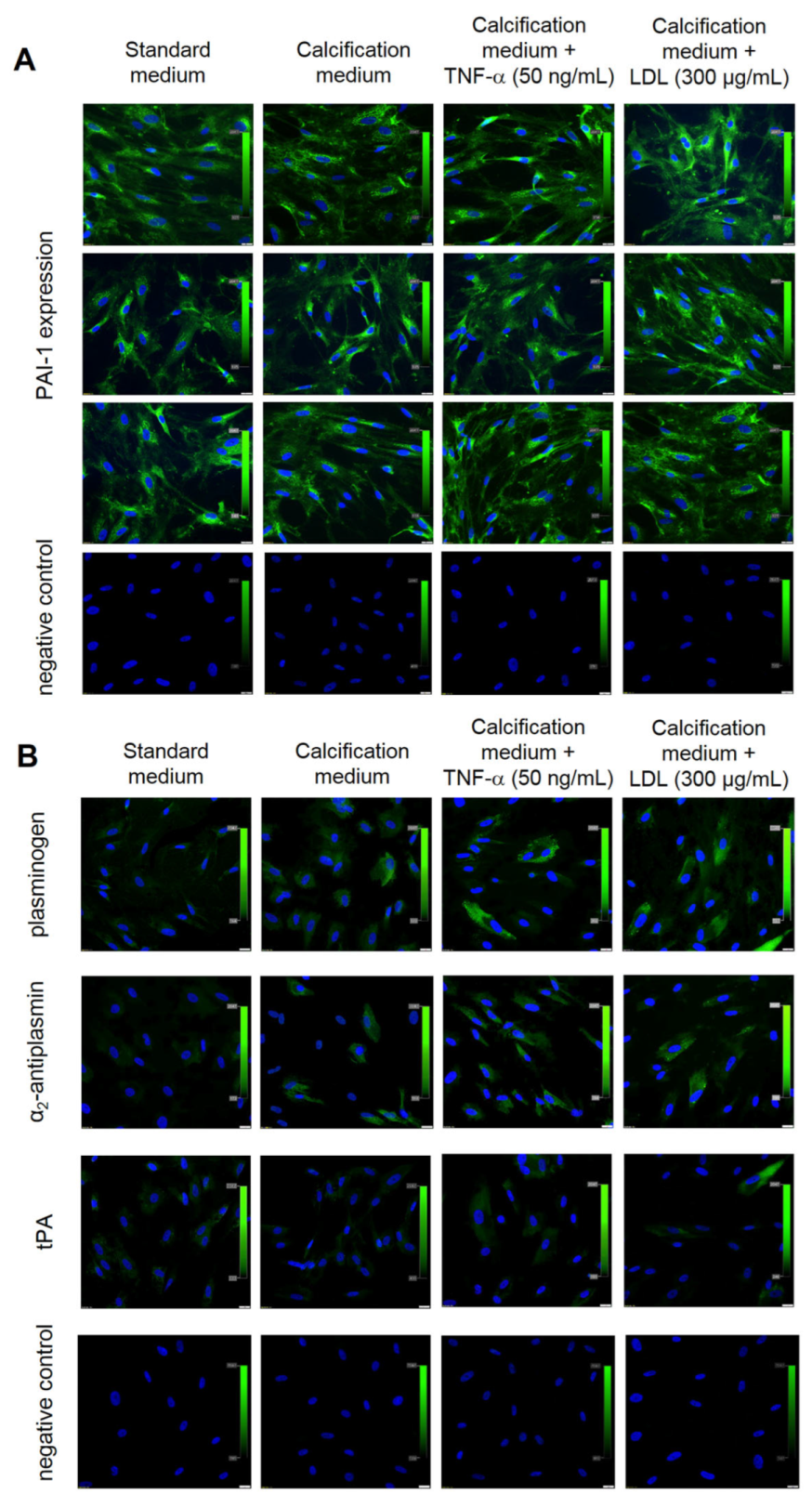
2.5. Valve Interstitial Cells In Vitro Cultures
2.6. CLT in VICs Supernatants
2.7. Relative Quantification of Transcripts by Real-Time PCR
2.8. Statistical Analyses
3. Results
3.1. Patients’ Characteristics
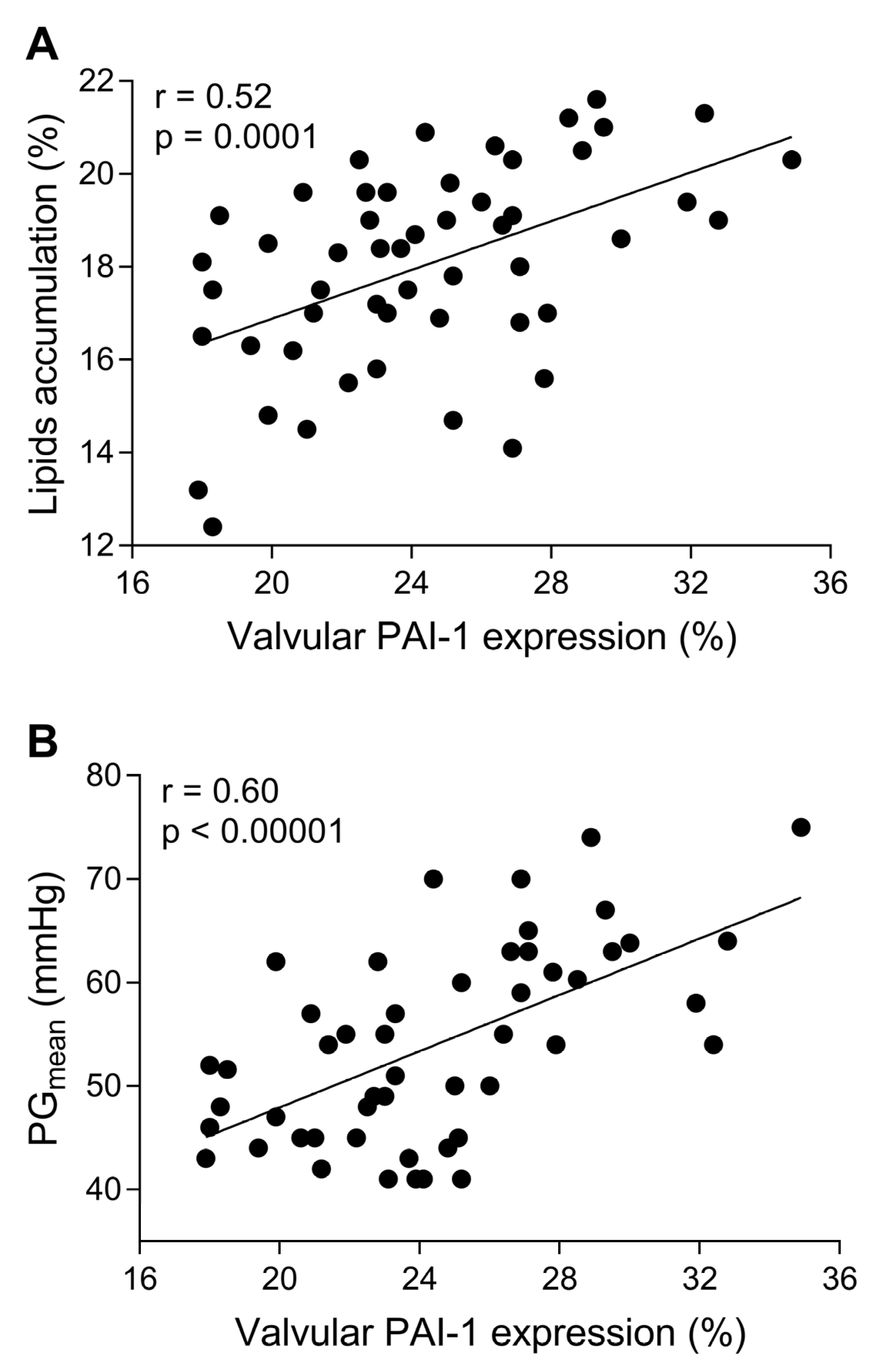
3.2. In Loco Studies
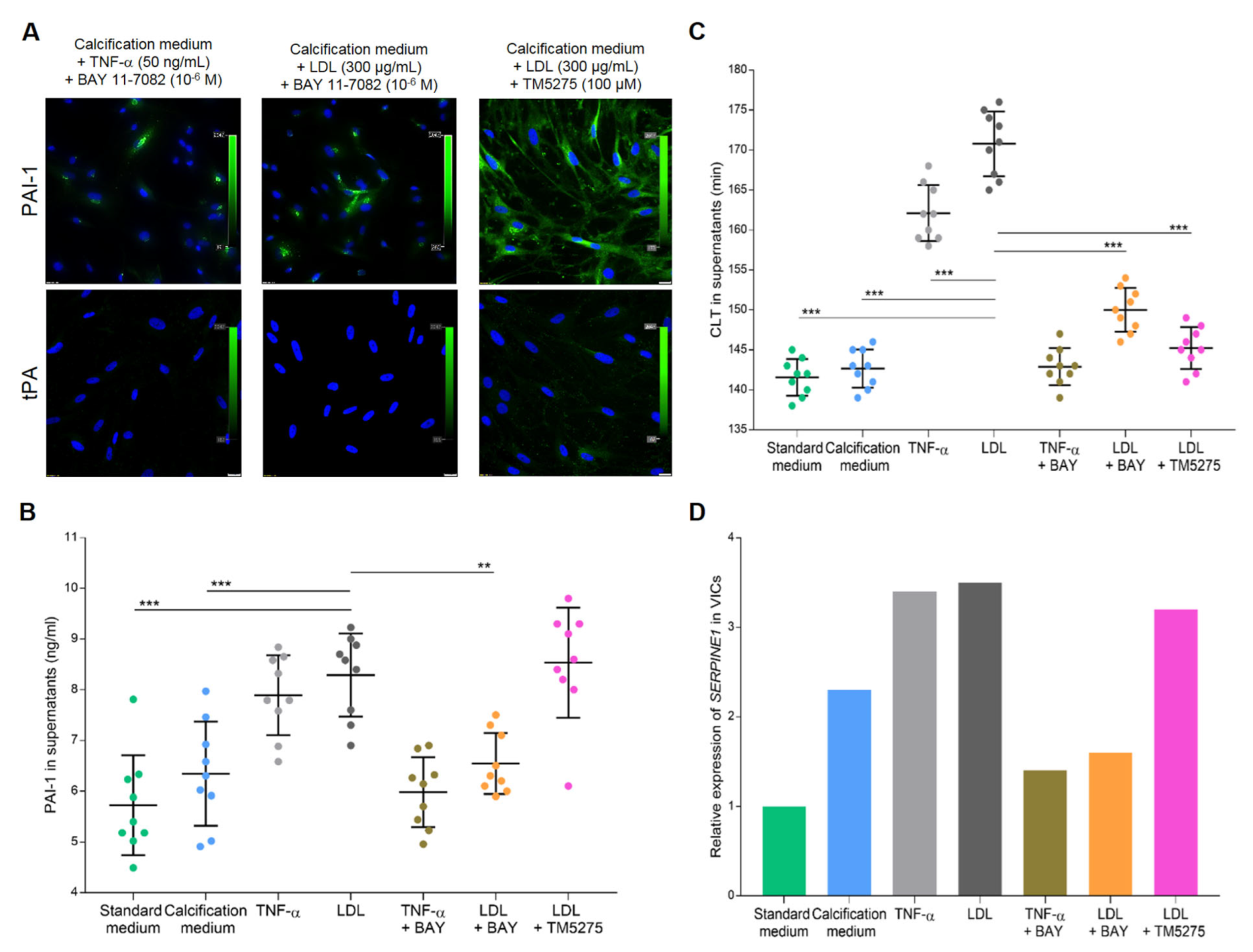
3.3. In Vitro Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindroos, M.; Kupari, M.; Heikkilä, J.; Tilvis, R. Prevalence of aortic valve abnormalities in the elderly: An echocardiographic study of a random population sample. J. Am. Coll. Cardiol. 1993, 21, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in Aortic Stenosis: The Skeleton Key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Natorska, J.; Undas, A. Blood coagulation and fibrinolysis in aortic valve stenosis: Links with inflammation and calcification. Thromb. Haemost. 2015, 114, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.C.; Joag, V.R.; Gotlieb, A.I. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol. 2007, 171, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Stubblefield, W.B.; Alves, N.J.; Rondina, M.T.; Kline, J.A. Variable Resistance to Plasminogen Activator Initiated Fibrinolysis for Intermediate-Risk Pulmonary Embolism. PLoS ONE 2016, 11, e0148747. [Google Scholar] [CrossRef]
- Natorska, J.; Wypasek, E.; Grudzień, G.; Sadowski, J.; Undas, A. Impaired fibrinolysis is associated with the severity of aortic stenosis in humans. J. Thromb. Haemost. 2013, 11, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Siudut, J.; Natorska, J.; Wypasek, E.; Wiewiórka, Ł.; Ostrowska-Kaim, E.; Wiśniowska-Śmiałek, S.; Plens, K.; Legutko, J.; Undas, A. Impaired Fibrinolysis in Patients with Isolated Aortic Stenosis is Associated with Enhanced Oxidative Stress. J. Clin. Med. 2020, 9, 2002. [Google Scholar] [CrossRef]
- Kochtebane, N.; Choqueux, C.; Passefort, S.; Nataf, P.; Messika-Zeitoun, D.; Bartagi, A.; Michel, J.-B.; Anglés-Cano, E.; Jacob, M.-P. Plasmin induces apoptosis of aortic valvular myofibroblasts. J. Pathol. 2010, 221, 37–48. [Google Scholar] [CrossRef]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between plasminogen activator inhibitor-1 and cardiovascular events: A systematic review and meta-analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.A.; Krause, M.P. PAI-1, the Plasminogen System, and Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 7066. [Google Scholar] [CrossRef]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen activator inhibitor-1 (PAI-1): A key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [PubMed]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kılıç, R.; Sarıkoç, A.; Piñol, R.; Hagl, S.; Lang, S.; et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kopytek, M.; Mazur, P.; Ząbczyk, M.; Undas, A.; Natorska, J. Diabetes concomitant to aortic stenosis is associated with increased expression of NF-κB and more pronounced valve calcification. Diabetologia 2021, 64, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- Siudut, J.; Natorska, J.; Wypasek, E.; Wiewiórka, Ł.; Ostrowska-Kaim, E.; Wiśniowska-Śmiałek, S.; Plens, K.; Musialek, P.; Legutko, J.; Undas, A. Apolipoproteins and lipoprotein(a) as factors modulating fibrin clot properties in patients with severe aortic stenosis. Atherosclerosis 2022, 344, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Vahanian, A.; Beyersdorf, F.; Praz, F.; Milojevic, M.; Baldus, S.; Bauersachs, J.; Capodanno, D.; Conradi, L.; De Bonis, M.; De Paulis, R.; et al. ESC/EACTS Scientific Document Group. 2021 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 2022, 43, 561–632. [Google Scholar] [CrossRef]
- Kopytek, M.; Ząbczyk, M.; Mazur, P.; Undas, A.; Natorska, J. Accumulation of advanced glycation end products (AGEs) is associated with the severity of aortic stenosis in patients with concomitant type 2 diabetes. Cardiovasc. Diabetol. 2020, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Ao, L.; Song, Y.; Babu, A.; Yang, X.; Wang, M.; Weyant, M.J.; Dinarello, C.A.; Cleveland, J.C., Jr.; Fullerton, D.A. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. Am. J. Physiol. Cell Physiol. 2008, 294, C29–C35. [Google Scholar] [CrossRef]
- Wypasek, E.; Natorska, J.; Mazur, P.; Kopytek, M.; Gawęda, B.; Kapusta, P.; Madeja, J.; Iwaniec, T.; Kapelak, B.; Undas, A. Effects of rivaroxaban and dabigatran on local expression of coagulation and inflammatory factors within human aortic stenotic valves. Vascul. Pharmacol. 2020, 130, 106679. [Google Scholar] [CrossRef] [PubMed]
- Gnanaguru, G.; Choi, A.R.; Amarnani, D.; D’Amore, P.A. Oxidized Lipoprotein Uptake Through the CD36 Receptor Activates the NLRP3 Inflammasome in Human Retinal Pigment Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4704–4712. [Google Scholar] [CrossRef]
- Yasui, H.; Suzuki, Y.; Sano, H.; Suda, T.; Chida, K.; Dan, T.; Miyata, T.; Urano, T. TM5275 prolongs secreted tissue plasminogen activator retention and enhances fibrinolysis on vascular endothelial cells. Thromb. Res. 2013, 132, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Matilla, L.; Roncal, C.; Ibarrola, J.; Arrieta, V.; García-Peña, A.; Fernández-Celis, A.; Navarro, A.; Álvarez, V.; Gainza, A.; Orbe, J.; et al. A Role for MMP-10 (Matrix Metalloproteinase-10) in Calcific Aortic Valve Stenosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1370–1382. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; Philippou, H.; Undas, A.; De Lange, Z.; Rijken, D.C.; Mutch, N.J. Subcommittee on Factor XIII and Fibrinogen, and the Subcommittee on Fibrinolysis. An international study on the feasibility of a standardized combined plasma clot turbidity and lysis assay: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Natorska, J.; Marek, G.; Hlawaty, M.; Sadowski, J.; Tracz, W.; Undas, A. Fibrin presence within aortic valves in patients with aortic stenosis: Association with in vivo thrombin generation and fibrin clot properties. Thromb. Haemost. 2011, 105, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Bolon, I.; Gazzeri, S.; Veyrenc, S.; Brambilla, C.; Brambilla, E. Expression of plasminogen activator inhibitors 1 and 2 in lung cancer and their role in tumor progression. Clin. Cancer Res. 1999, 5, 2094–2102. [Google Scholar] [PubMed]
- D’Auria, F.; Centurione, L.; Centurione, M.A.; Angelini, A.; Di Pietro, R. Tumor Necrosis Factor Related Apoptosis Inducing Ligand (Trail) in endothelial response to biomechanical and biochemical stresses in arteries. J. Cell. Biochem. 2015, 116, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Eren, M.; Painter, C.A.; Gleaves, L.A.; Schoenhard, J.A.; Atkinson, J.B.; Brown, N.J.; Vaughan, D.E. Tissue- and agonist-specific regulation of human and murine plasminogen activator inhibitor-1 promoters in transgenic mice. J. Thromb. Haemost. 2003, 1, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, N.; Katritsis, D.; Raggi, P. Visceral adipose tissue as a source of inflammation and promoter of atherosclerosis. Atherosclerosis 2014, 233, 104–112. [Google Scholar] [CrossRef]
- Khoukaz, H.B.; Ji, Y.; Braet, D.J.; Vadali, M.; Abdelhamid, A.A.; Emal, C.D.; Lawrence, D.; Fay, W.P. Drug Targeting of Plasminogen Activator Inhibitor-1 Inhibits Metabolic Dysfunction and Atherosclerosis in a Murine Model of Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1479–1490. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | AS Patients (n = 75) |
|---|---|
| Age, years | 66 [60–71] |
| Male, n (%) | 46 (61.3) |
| BMI, kg m−2 | 28 [25.7–30.6] |
| Risk factors, n (%) | |
| Arterial hypertension | 67 (89.3) |
| Hypercholesterolemia | 64 (85.3) |
| Current smoking | 13 (17.3) |
| Medications, n (%) | |
| Beta-blockers | 54 (72) |
| Acetylsalicylic acid | 51 (68) |
| ACE inhibitors | 48 (64) |
| Statins | 57 (76) |
| Rivaroxaban | 5 (6.7) |
| Apixaban | 3 (4) |
| Dabigatran | 4 (5.3) |
| Echocardiographic parameters | |
| Mean gradient, mmHg | 50 [44–58] |
| Maximal gradient, mmHg | 82 [74–94] |
| AVA, cm2 | 0.8 [0.7–0.9] |
| LVEF, % | 60 [55–65] |
| Laboratory investigations | |
| Fibrinogen, g/L | 3.4 ± 0.74 |
| Creatinine, µmol/L | 76 [70–92] |
| CRP, mg/L | 2.0 [1.0–4.0] |
| Glucose, mmol/L | 5.4 [5.0–5.6] |
| Total cholesterol mmol/L | 4.1 [3.5–4.8] |
| LDL cholesterol, mmol/L | 2.5 [2.0–3.3] |
| HDL cholesterol, mmol/L | 1.6 [1.3–1.7] |
| Triglycerides, mmol/L | 1.1 [0.9–1.7] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopytek, M.; Ząbczyk, M.; Mazur, P.; Undas, A.; Natorska, J. PAI-1 Overexpression in Valvular Interstitial Cells Contributes to Hypofibrinolysis in Aortic Stenosis. Cells 2023, 12, 1402. https://doi.org/10.3390/cells12101402
Kopytek M, Ząbczyk M, Mazur P, Undas A, Natorska J. PAI-1 Overexpression in Valvular Interstitial Cells Contributes to Hypofibrinolysis in Aortic Stenosis. Cells. 2023; 12(10):1402. https://doi.org/10.3390/cells12101402
Chicago/Turabian StyleKopytek, Magdalena, Michał Ząbczyk, Piotr Mazur, Anetta Undas, and Joanna Natorska. 2023. "PAI-1 Overexpression in Valvular Interstitial Cells Contributes to Hypofibrinolysis in Aortic Stenosis" Cells 12, no. 10: 1402. https://doi.org/10.3390/cells12101402
APA StyleKopytek, M., Ząbczyk, M., Mazur, P., Undas, A., & Natorska, J. (2023). PAI-1 Overexpression in Valvular Interstitial Cells Contributes to Hypofibrinolysis in Aortic Stenosis. Cells, 12(10), 1402. https://doi.org/10.3390/cells12101402