
Mitochondrial Transfer to Host Cells from Ex Vivo Expanded Donor Hematopoietic Stem Cells

 , and
, and 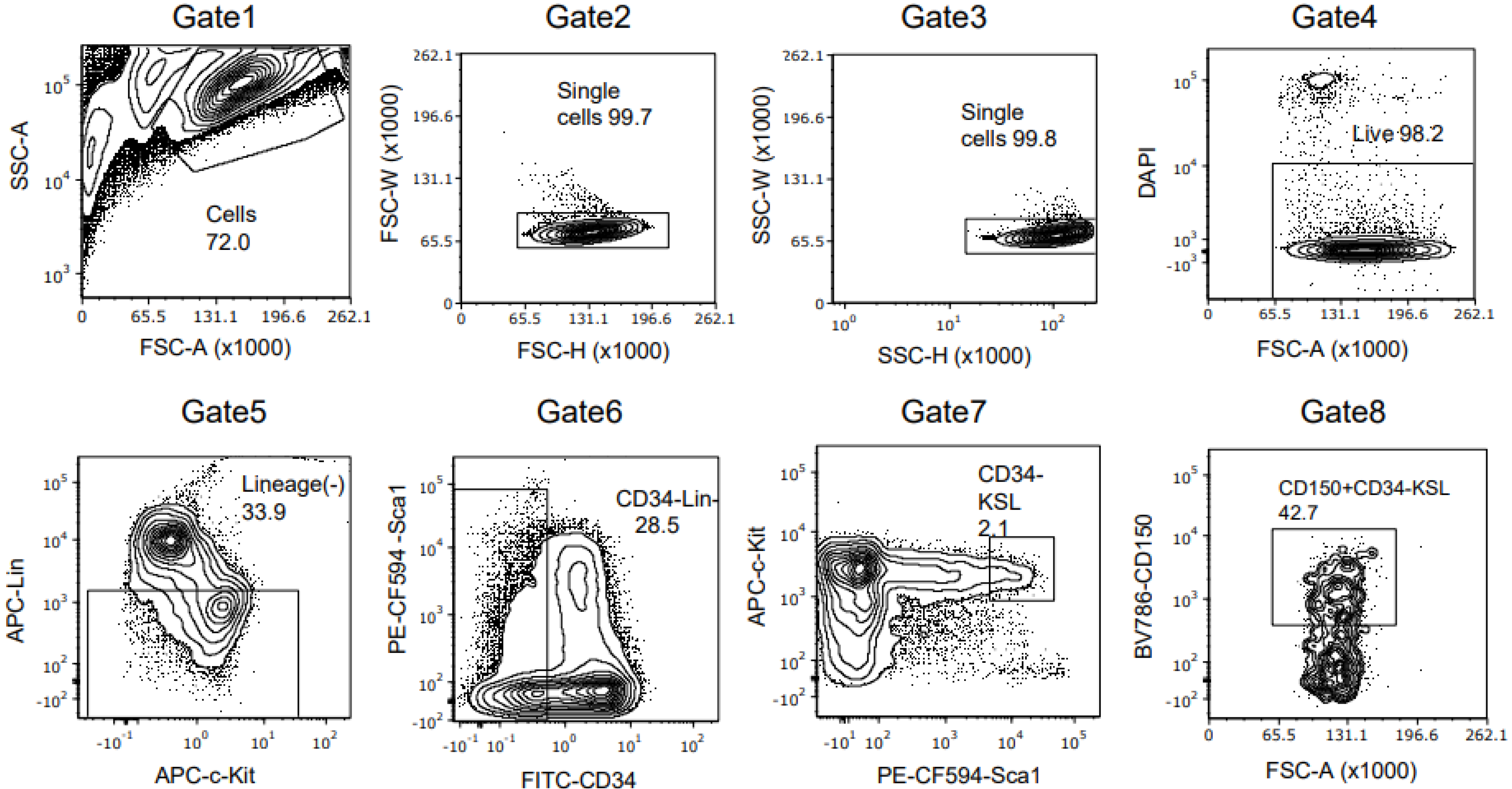{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Mice
2.3. Complete Blood Counts
2.4. Light Microscopy
2.5. Flow Cytometry
2.6. HSC Isolation by Fluorescence-Activated Cell Sorting
2.7. HSC Ex Vivo Culture
2.8. Quality Tests of Cultured Cells
2.9. Transplant Assays
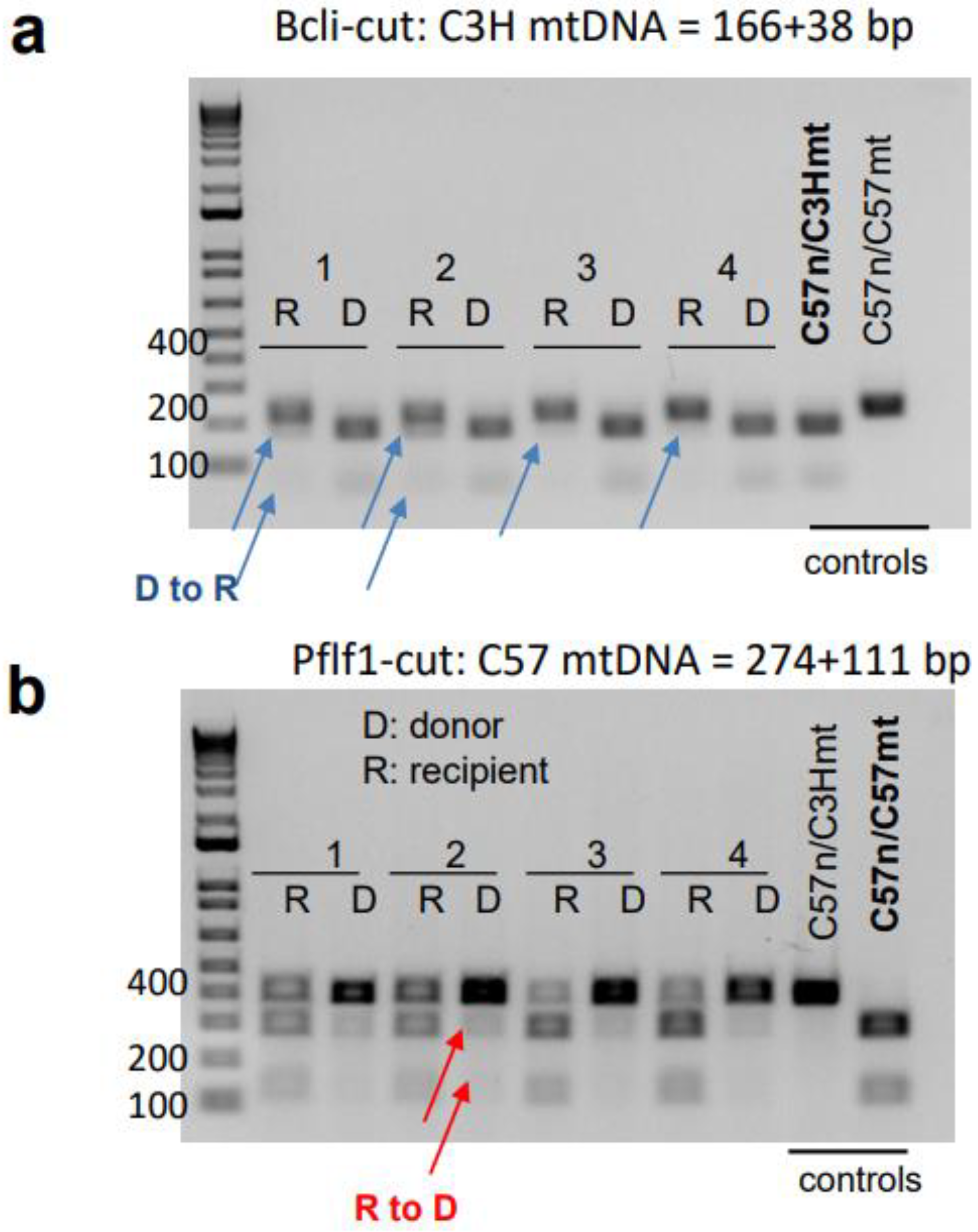
2.10. Detection of C3H and C57 mtDNA
2.11. Statistical Analysis
3. Results
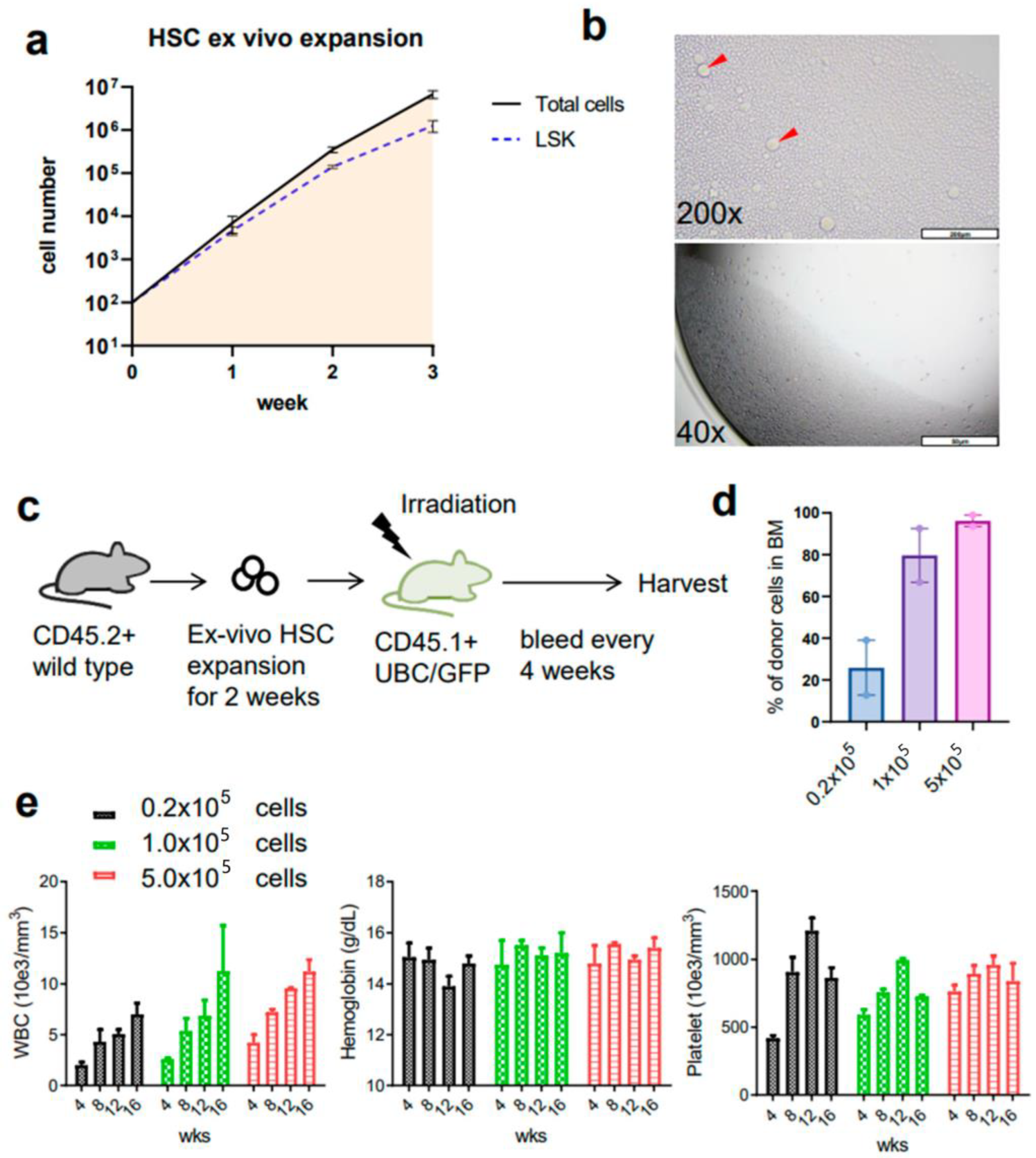
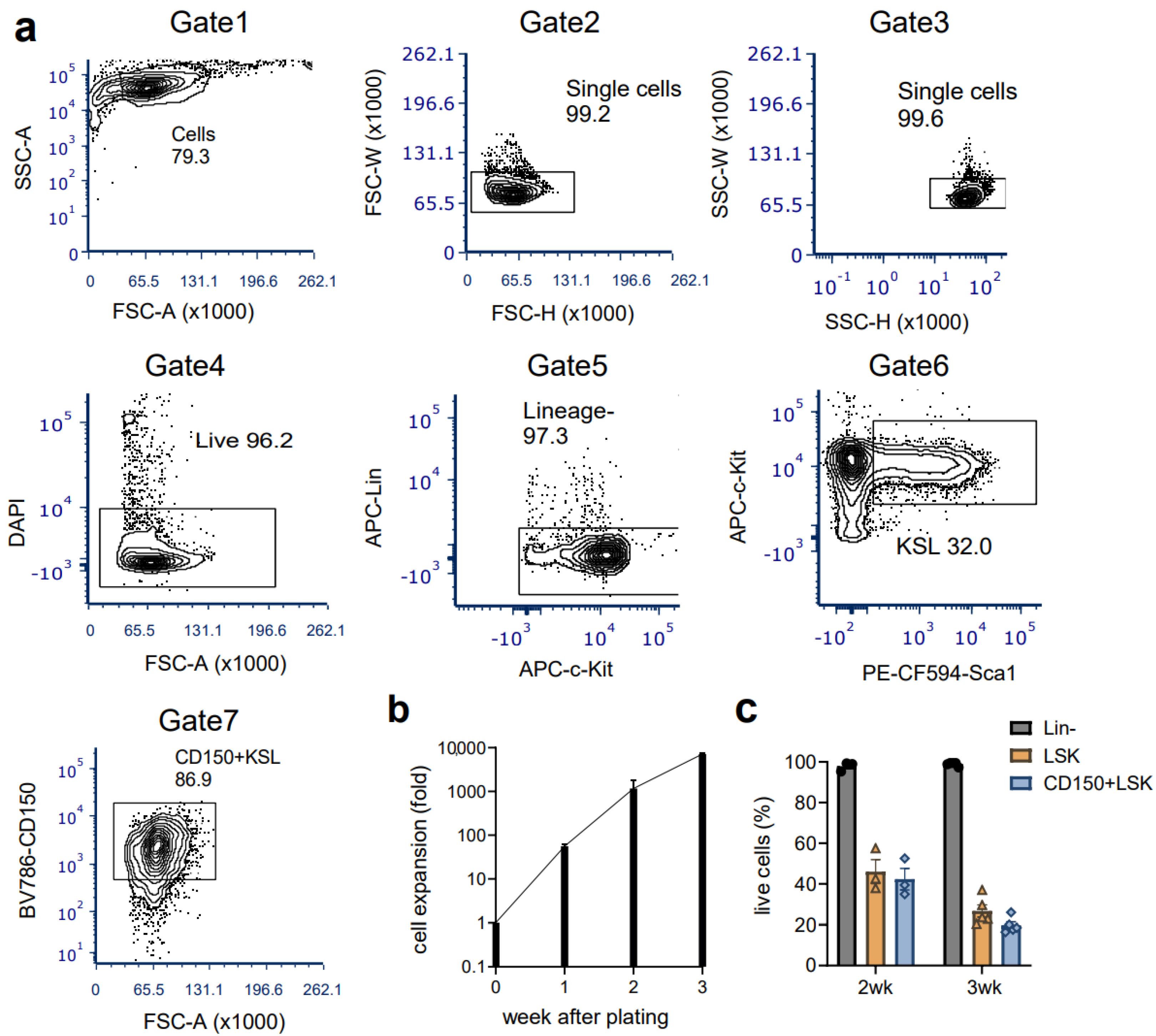
3.1. Ex Vivo Culture of Mouse HSCs from Young Mice Reveals Robust Expansion Pattern and Reserved Functional HSCs in the Transplant Assay
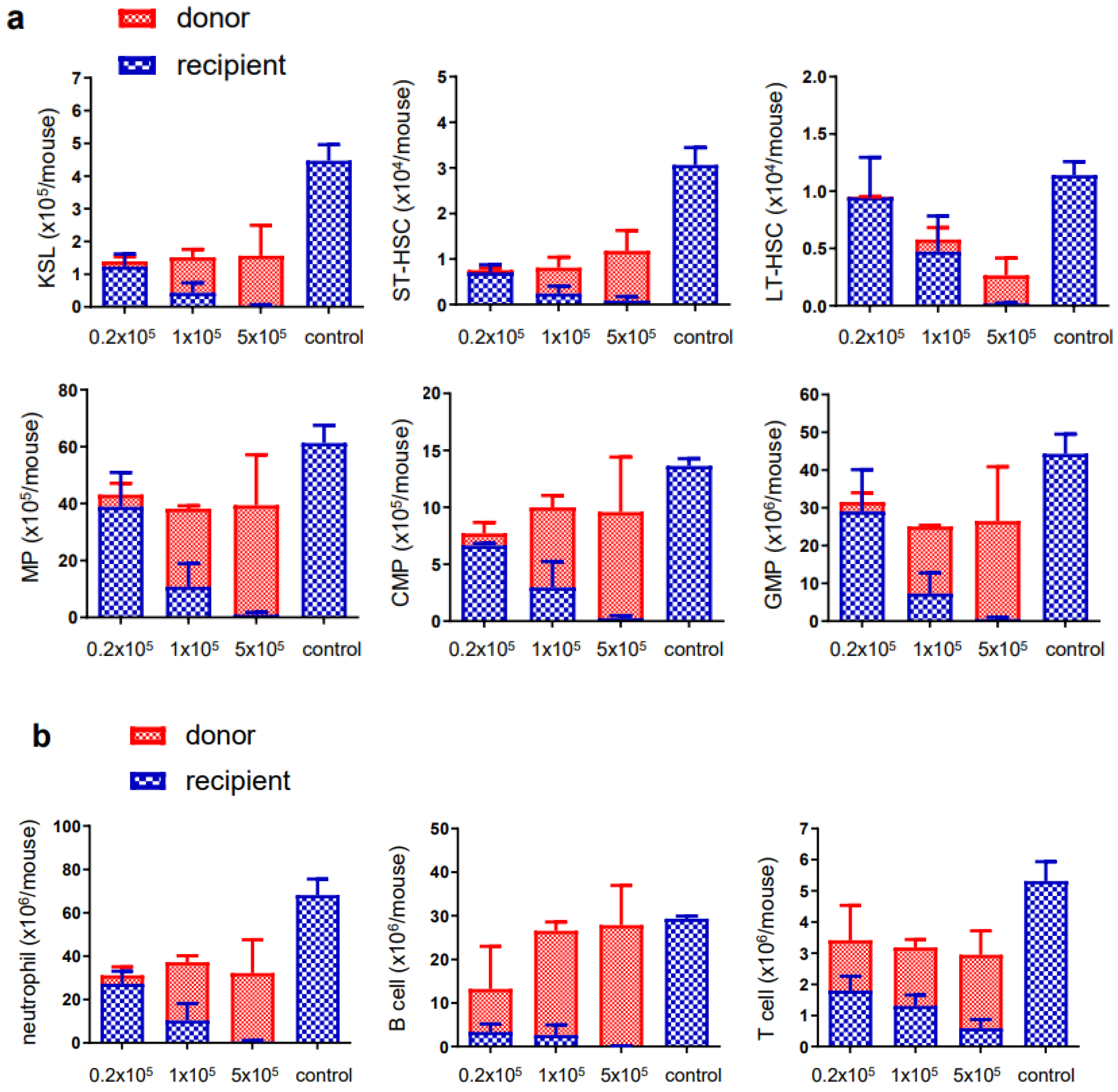
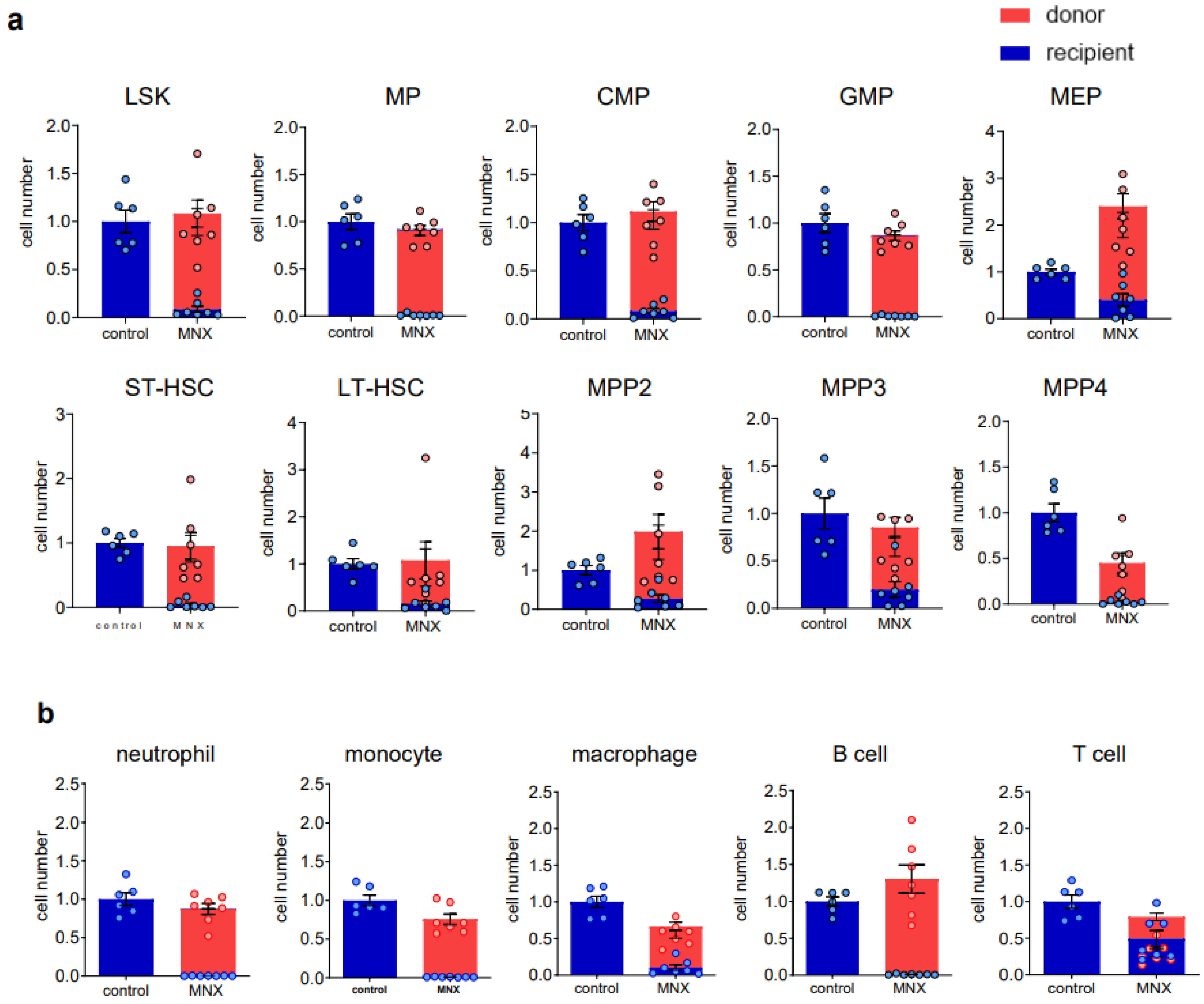
3.2. Ex Vivo Expanded Mouse HSCs Efficiently Replaced Host Hematopoietic Progenitor Cells (HPCs) or HSCs in a Donor Cell Number-Dependent Manner
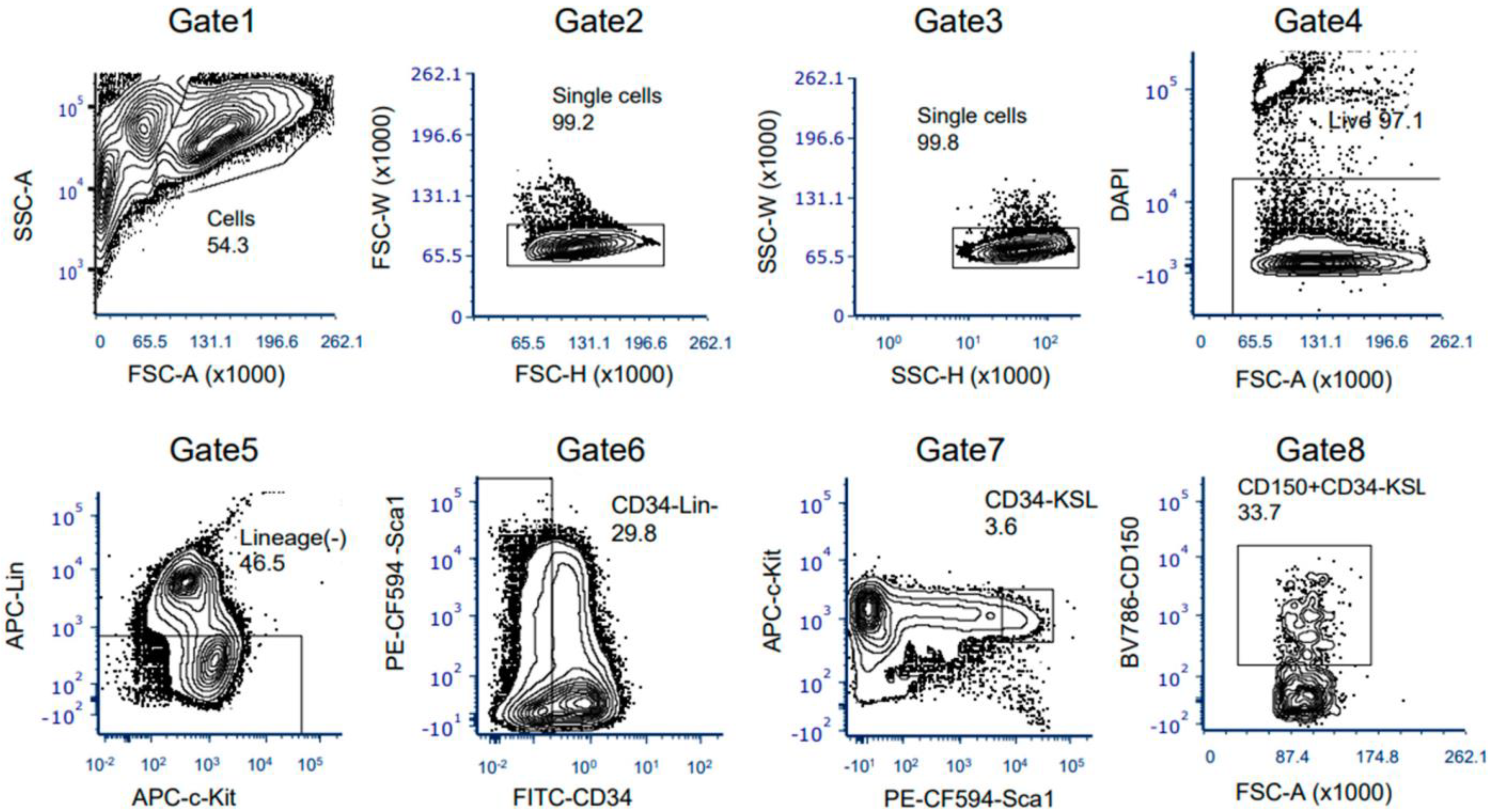
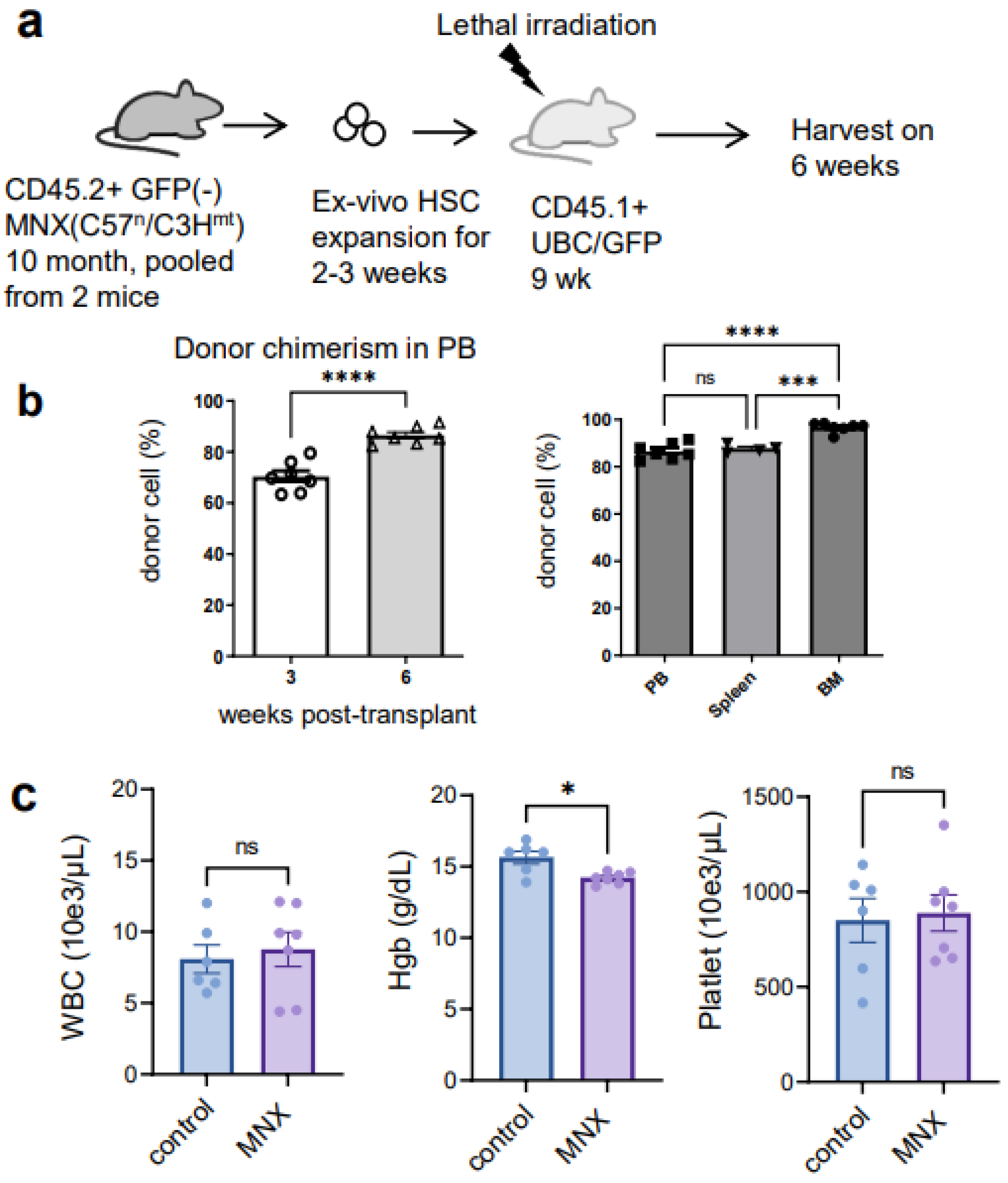
3.3. The Ex Vivo HSC Culture from MNX Mice Showed a Comparable Level of CD150+ HSCs with Potential of Engraftment after Transplantation
3.4. mtDNA Transfer from Donor to Host in a Transplant Model Utilizing Ex Vivo Expanded Mouse HSC Culture
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- da Silveira, W.A.; Fazelinia, H.; Rosenthal, S.B.; Laiakis, E.C.; Kim, M.S.; Meydan, C.; Kidane, Y.; Rathi, K.S.; Smith, S.M.; Stear, B.; et al. Comprehensive Multi-omics Analysis Reveals Mitochondrial Stress as a Central Biological Hub for Spaceflight Impact. Cell 2020, 183, 1185–1201.e20. [Google Scholar] [CrossRef]
- Geto, Z.; Molla, M.D.; Challa, F.; Belay, Y.; Getahun, T. Mitochondrial Dynamic Dysfunction as a Main Triggering Factor for Inflammation Associated Chronic Non-Communicable Diseases. J. Inflamm. Res. 2020, 13, 97–107. [Google Scholar] [CrossRef]
- Giang, A.-H.; Raymond, T.; Brookes, P.; de Mesy Bentley, K.; Schwarz, E.; O’Keefe, R.; Eliseev, R. Mitochondrial Dysfunction and Permeability Transition in Osteosarcoma Cells Showing the Warburg Effect. J. Biol. Chem. 2013, 288, 33303–33311. [Google Scholar] [CrossRef]
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Klopstock, T.; Kornblum, C.; Mancuso, M.; McFarland, R.; Sue, C.M.; Suomalainen, A.; Taylor, R.W.; et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021, 20, 573–584. [Google Scholar] [CrossRef] [PubMed]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Votyakova, T.V.; Reynolds, I.J.; Mathews, C.E. Nuclear and Mitochondrial Interaction Involving mt-Nd2 Leads to Increased Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2007, 282, 5171–5179. [Google Scholar] [CrossRef]
- Kesterson, R.A.; Johnson, L.W.; Lambert, L.J.; Vivian, J.L.; Welch, D.R.; Ballinger, S.W. Generation of Mitochondrial-nuclear eXchange Mice via Pronuclear Transfer. Bio. Protoc. 2016, 6, e1976. [Google Scholar] [CrossRef]
- Brinker, A.E.; Vivian, C.J.; Beadnell, T.C.; Koestler, D.C.; Teoh, S.T.; Lunt, S.Y.; Welch, D.R. Mitochondrial Haplotype of the Host Stromal Microenvironment Alters Metastasis in a Non-cell Autonomous Manner. Cancer Res 2020, 80, 1118–1129. [Google Scholar] [CrossRef]
- Beadnell, T.; Fain, C.; Vivian, C.; King, J.; Hastings, R.; Markiewicz, M.; Welch, D. Mitochondrial genetics cooperate with nuclear genetics to selectively alter immune cell development/trafficking. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165648. [Google Scholar] [CrossRef] [PubMed]
- Vivian, C.J.; Brinker, A.E.; Graw, S.; Koestler, D.C.; Legendre, C.; Gooden, G.C.; Salhia, B.; Welch, D.R. Mitochondrial Genomic Backgrounds Affect Nuclear DNA Methylation and Gene Expression. Cancer Res. 2017, 77, 6202–6214. [Google Scholar] [CrossRef]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.d.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Konari, N.; Nagaishi, K.; Kikuchi, S.; Fujimiya, M. Mitochondria transfer from mesenchymal stem cells structurally and functionally repairs renal proximal tubular epithelial cells in diabetic nephropathy in vivo. Sci. Rep. 2019, 9, 5184. [Google Scholar] [CrossRef]
- Liu, K.; Guo, L.; Zhou, Z.; Pan, M.; Yan, C. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc. Res. 2019, 123, 74–80. [Google Scholar] [CrossRef]
- Osawa, M.; Hanada, K.-I.; Hamada, H.; Nakauchi, H. Long-Term Lymphohematopoietic Reconstitution by a Single CD34-Low/Negative Hematopoietic Stem Cell. Science 1996, 273, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019, 571, 117–121. [Google Scholar] [CrossRef]
- Wilkinson, A.C.; Ishida, R.; Nakauchi, H.; Yamazaki, S. Long-term ex vivo expansion of mouse hematopoietic stem cells. Nat. Protoc. 2020, 15, 628–648. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Yamazaki, S.; Wilkinson, A.C.; Nakauchi, H.; Nakamura, Y. Polyvinyl alcohol hydrolysis rate and molecular weight influence human and murine HSC activity ex vivo. Stem Cell Res. 2021, 56, 102531. [Google Scholar] [CrossRef]
- Suzuki, T.; Ishii, S.; Shinohara, M.; Kawano, Y.; Wakahashi, K.; Kawano, H.; Sada, A.; Minagawa, K.; Hamada, M.; Takahashi, S.; et al. Mobilization efficiency is critically regulated by fat via marrow PPARδ. Haematologica 2021, 106, 1671–1683. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Zelickson, B.R.; Johnson, L.W.; Moellering, D.; Westbrook, D.G.; Pompilius, M.; Sammy, M.; Johnson, M.; Dunham-Snary, K.; Cao, X.; et al. Mitochondrial genetic background modulates bioenergetics and susceptibility to acute cardiac volume overload. Biochem. J. 2013, 455, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Guichard, J.L.; Kane, M.S.; Grenett, M.; Sandel, M.; Benavides, G.A.; Bradley, W.; Powell, P.C.; Darley-Usmar, V.; Ballinger, S.W.; Dell’Italia, L.J. Mitochondrial Haplotype Modulates Genome Expression and Mitochondrial Structure/Function in Cardiomyocytes following Volume Overload. Am. J. Physiol. Circ. Physiol. 2023, 324, H484–H493. [Google Scholar] [CrossRef]
- Frisch, B.J.; Hoffman, C.M.; Latchney, S.E.; LaMere, M.W.; Myers, J.; Ashton, J.; Li, A.J.; Saunders, J.; Palis, J.; Perkins, A.S.; et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via interleukin-1B. J. Clin. Investig. 2019, 5, e124213. [Google Scholar] [CrossRef]
- Balderman, S.R.; Li, A.J.; Hoffman, C.M.; Frisch, B.J.; Goodman, A.N.; LaMere, M.W.; Georger, M.A.; Evans, A.G.; Liesveld, J.L.; Becker, M.W.; et al. Targeting of the bone marrow microenvironment improves outcome in a murine model of myelodysplastic syndrome. Blood 2016, 127, 616–625. [Google Scholar] [CrossRef]
- Zijlmans, J.M.J.M.; Visser, J.W.M.; Laterveer, L.; Kleiverda, K.; Heemskerk, D.P.M.; Kluin, P.M.; Willemze, R.; Fibbe, W.E. The early phase of engraftment after murine blood cell transplantation is mediated by hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1998, 95, 725–729. [Google Scholar] [CrossRef]
- Kobayashi, H.; Morikawa, T.; Okinaga, A.; Hamano, F.; Hashidate-Yoshida, T.; Watanuki, S.; Hishikawa, D.; Shindou, H.; Arai, F.; Kabe, Y.; et al. Environmental Optimization Enables Maintenance of Quiescent Hematopoietic Stem Cells Ex Vivo. Cell Rep. 2019, 28, 145–158.e9. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2022, 17, 98–106. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Mok, B.Y.; De Moraes, M.H.; Zeng, J.; Yeh, M.M.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef]
- Greenfield, A.; Braude, P.; Flinter, F.; Lovell-Badge, R.; Ogilvie, C.; Perry, A.C.F. Assisted reproductive technologies to prevent human mitochondrial disease transmission. Nat. Biotechnol. 2017, 35, 1059–1068. [Google Scholar] [CrossRef]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef]
- Maeda, H.; Kami, D.; Maeda, R.; Shikuma, A.; Gojo, S. Generation of somatic mitochondrial DNA-replaced cells for mitochondrial dysfunction treatment. Sci. Rep. 2021, 11, 10897. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Golan, K.; Singh, A.K.; Kollet, O.; Bertagna, M.; Althoff, M.J.; Khatib-Massalha, E.; Petrovich-Kopitman, E.; Wellendorf, A.M.; Massalha, H.; Levin-Zaidman, S.; et al. Bone marrow regeneration requires mitochondrial transfer from donor Cx43-expressing hematopoietic progenitors to stroma. Blood 2020, 136, 2607–2619. [Google Scholar] [CrossRef]
- Pham, A.H.; McCaffery, J.M.; Chan, D.C. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 2012, 50, 833–843. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawano, H.; Kawano, Y.; Yu, C.; LaMere, M.W.; McArthur, M.J.; Becker, M.W.; Ballinger, S.W.; Gojo, S.; Eliseev, R.A.; Calvi, L.M. Mitochondrial Transfer to Host Cells from Ex Vivo Expanded Donor Hematopoietic Stem Cells. Cells 2023, 12, 1473. https://doi.org/10.3390/cells12111473
Kawano H, Kawano Y, Yu C, LaMere MW, McArthur MJ, Becker MW, Ballinger SW, Gojo S, Eliseev RA, Calvi LM. Mitochondrial Transfer to Host Cells from Ex Vivo Expanded Donor Hematopoietic Stem Cells. Cells. 2023; 12(11):1473. https://doi.org/10.3390/cells12111473
Chicago/Turabian StyleKawano, Hiroki, Yuko Kawano, Chen Yu, Mark W. LaMere, Matthew J. McArthur, Michael W. Becker, Scott W. Ballinger, Satoshi Gojo, Roman A. Eliseev, and Laura M. Calvi. 2023. "Mitochondrial Transfer to Host Cells from Ex Vivo Expanded Donor Hematopoietic Stem Cells" Cells 12, no. 11: 1473. https://doi.org/10.3390/cells12111473
APA StyleKawano, H., Kawano, Y., Yu, C., LaMere, M. W., McArthur, M. J., Becker, M. W., Ballinger, S. W., Gojo, S., Eliseev, R. A., & Calvi, L. M. (2023). Mitochondrial Transfer to Host Cells from Ex Vivo Expanded Donor Hematopoietic Stem Cells. Cells, 12(11), 1473. https://doi.org/10.3390/cells12111473