

Microphysiological Models for Mechanistic-Based Prediction of Idiosyncratic DILI


Abstract
:1. Introduction
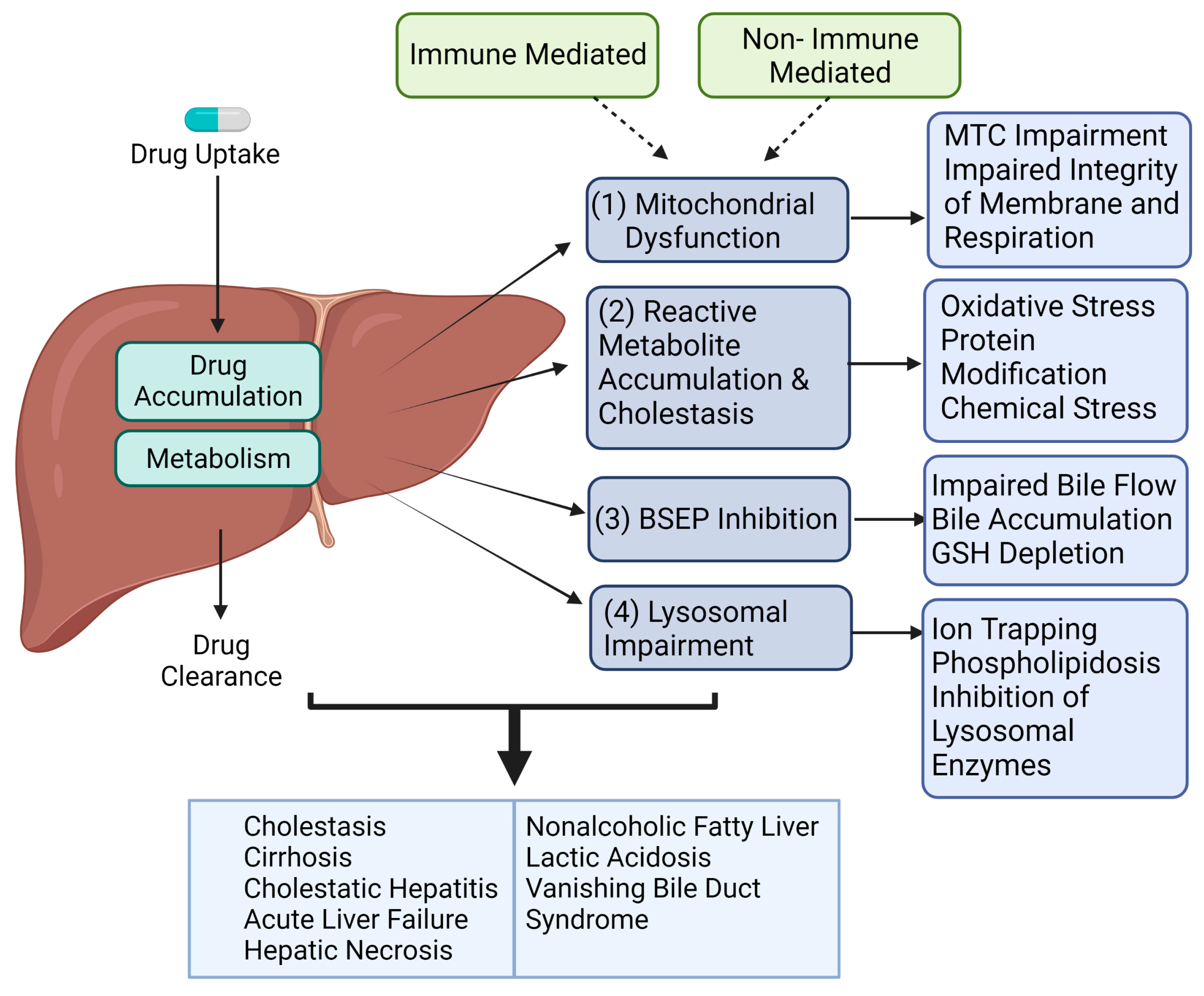
2. Mechanism of DILI in Humans
2.1. Immune-Mediated Mechanism
2.2. Reactive Metabolite Accumulation & Cholestasis
2.3. Mitochondrial Dysfunction
2.4. BSEP Inhibition
2.5. Lysosomal Impairment
2.6. Regulatory Considerations for DILI per Guidance for Industry
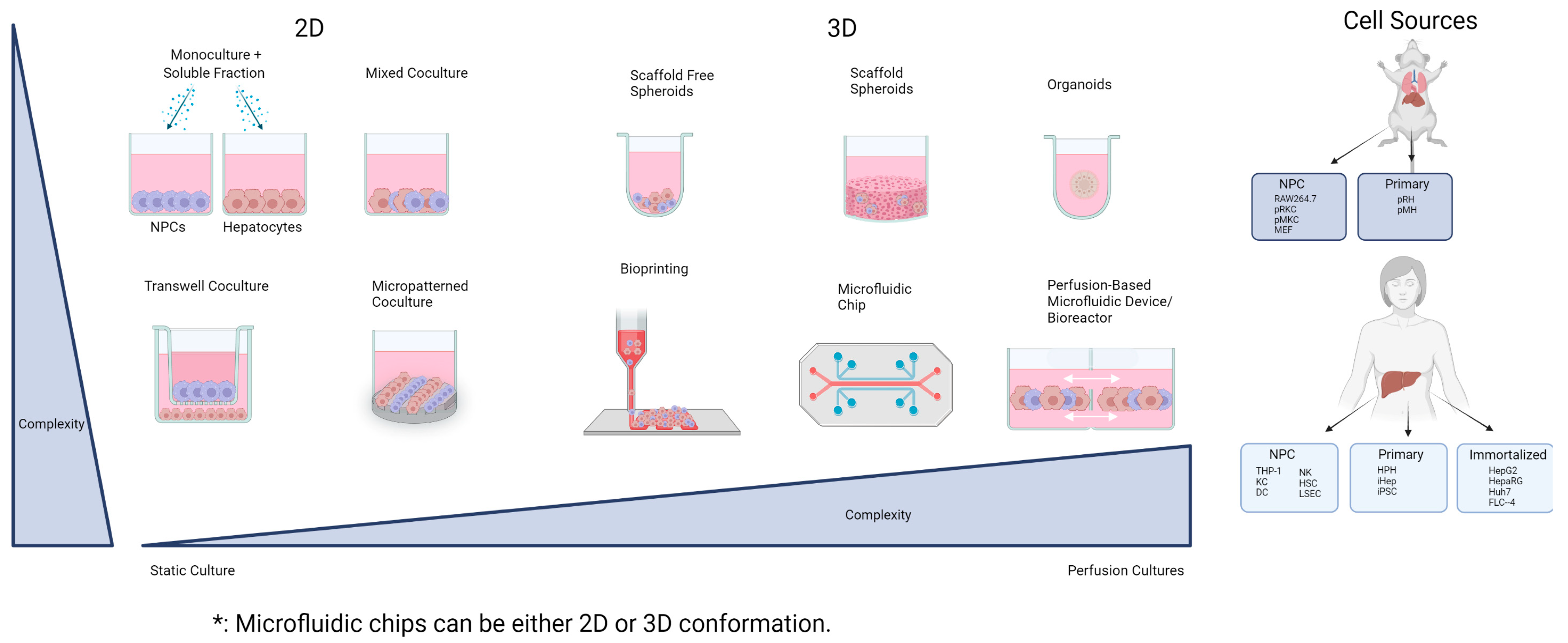
3. Overview of Hepatocyte–NPC Co-Culture Models
3.1. Two-Dimensional Co-Cultures
3.1.1. Mixed Co-Cultures
3.1.2. Micropatterned Culture
3.1.3. Transwell
3.2. Three-Dimensional Co-Cultures
3.2.1. Spheroid NPC Co-Cultures
3.2.2. iPSC-Derived Liver Organoids
3.2.3. Bioprinting
3.2.4. Perfusion-Based Co-Culture Models
Bioreactors
Microfluidic Systems (Liver-on-a-Chip)
3.3. Challenges Associated with Establishing Hepatocyte–NPC Co-Culture Models for DILI
4. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- FDA, T.U. Guidance for Industry Drug-Induced Liver Injury: Premarketing Clinical Evaluation. Available online: https://www.fda.gov/media/116737/download (accessed on 18 May 2023).
- Bjornsson, E.S.; Bergmann, O.M.; Bjornsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013, 144, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Hunt, C.M.; Suzuki, A.; Papay, J.I.; Beach, K.J.; Cheetham, T.C. Characterizing phenotypes and outcomes of drug-associated liver injury using electronic medical record data. Pharmacoepidemiol. Drug Saf. 2013, 22, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Sgro, C.; Clinard, F.; Ouazir, K.; Chanay, H.; Allard, C.; Guilleminet, C.; Lenoir, C.; Lemoine, A.; Hillon, P. Incidence of drug-induced hepatic injuries: A French population-based study. Hepatology 2002, 36, 451–455. [Google Scholar] [CrossRef]
- Fontana, R.J.; Hayashi, P.H.; Gu, J.; Reddy, K.R.; Barnhart, H.; Watkins, P.B.; Serrano, J.; Lee, W.M.; Chalasani, N.; Stolz, A.; et al. Idiosyncratic drug-induced liver injury is associated with substantial morbidity and mortality within 6 months from onset. Gastroenterology 2014, 147, 96–108.e4. [Google Scholar] [CrossRef]
- Ostapowicz, G.; Fontana, R.J.; Schiodt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef]
- Reuben, A.; Koch, D.G.; Lee, W.M.; The Acute Liver Failure Study Group. Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef]
- Hughes, B. Industry concern over EU hepatotoxicity guidance. Nat. Rev. Drug Discov. 2008, 7, 719. [Google Scholar] [CrossRef]
- Greek, R.; Menache, A. Systematic reviews of animal models: Methodology versus epistemology. Int. J. Med. Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef]
- Uetrecht, J. Mechanistic Studies of Idiosyncratic DILI: Clinical Implications. Front. Pharmacol. 2019, 10, 837. [Google Scholar] [CrossRef]
- Ng, W.; Lobach, A.R.; Zhu, X.; Chen, X.; Liu, F.; Metushi, I.G.; Sharma, A.; Li, J.; Cai, P.; Ip, J.; et al. Animal models of idiosyncratic drug reactions. Adv. Pharmacol. 2012, 63, 81–135. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Jaeschke, H. Animal models of drug-induced liver injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, W.; Kappenberg, F.; Brecklinghaus, T.; Stoeber, R.; Marchan, R.; Zhang, M.; Ebbert, K.; Kirschner, H.; Grinberg, M.; Leist, M.; et al. Prediction of human drug-induced liver injury (DILI) in relation to oral doses and blood concentrations. Arch. Toxicol. 2019, 93, 1609–1637. [Google Scholar] [CrossRef] [PubMed]
- Gulmez, S.E.; Larrey, D.; Pageaux, G.P.; Lignot, S.; Lassalle, R.; Jove, J.; Gatta, A.; McCormick, P.A.; Metselaar, H.J.; Monteiro, E.; et al. Transplantation for acute liver failure in patients exposed to NSAIDs or paracetamol (acetaminophen): The multinational case-population SALT study. Drug Saf. 2013, 36, 135–144. [Google Scholar] [CrossRef]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiodt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef]
- Daly, A.K.; Day, C.P. Genetic association studies in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 116–126. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Nicoletti, P.; Abramson, K.; Andrade, R.J.; Bjornsson, E.S.; Chalasani, N.; Fontana, R.J.; Hallberg, P.; Li, Y.J.; Lucena, M.I.; et al. A Missense Variant in PTPN22 is a Risk Factor for Drug-induced Liver Injury. Gastroenterology 2019, 156, 1707–1716.e2. [Google Scholar] [CrossRef]
- Aithal, G.P.; Ramsay, L.; Daly, A.K.; Sonchit, N.; Leathart, J.B.; Alexander, G.; Kenna, J.G.; Caldwell, J.; Day, C.P. Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology 2004, 39, 1430–1440. [Google Scholar] [CrossRef]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.; Lee, W.; Stolz, A.; Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.; Barnhart, H.; et al. Features and Outcomes of 899 Patients with Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology 2015, 148, 1340–1352.e7. [Google Scholar] [CrossRef]
- Benichou, C. Criteria of drug-induced liver disorders. Report of an international consensus meeting. J. Hepatol. 1990, 11, 272–276. [Google Scholar] [CrossRef]
- Kaplowitz, N. Drug-induced liver injury. Clin. Infect. Dis. 2004, 38 (Suppl. S2), S44–S48. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.P.; Cheng, L.; Ju, C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 2008, 84, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Han, J.; Lee, J.; Lee, A.; Cho, S.; Rho, P.; Kang, M.W.; Jang, J.; Jung, E.; Choi, J.; et al. Intrahepatic infiltration of activated CD8+ T cells and mononuclear phagocyte is associated with idiosyncratic drug-induced liver injury. Front Immunol. 2023, 14, 1138112. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Senior, J.R. Recognizing drug-induced liver injury: Current problems, possible solutions. Toxicol. Pathol. 2005, 33, 155–164. [Google Scholar] [CrossRef]
- Lammert, C.; Einarsson, S.; Saha, C.; Niklasson, A.; Bjornsson, E.; Chalasani, N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: Search for signals. Hepatology 2008, 47, 2003–2009. [Google Scholar] [CrossRef]
- Adams, D.H.; Ju, C.; Ramaiah, S.K.; Uetrecht, J.; Jaeschke, H. Mechanisms of immune-mediated liver injury. Toxicol. Sci. 2010, 115, 307–321. [Google Scholar] [CrossRef]
- Liaskou, E.; Wilson, D.V.; Oo, Y.H. Innate immune cells in liver inflammation. Mediat. Inflamm. 2012, 2012, 949157. [Google Scholar] [CrossRef]
- Waddington, J.C.; Meng, X.; Naisbitt, D.J.; Park, K. Immune drug-induced liver disease and drugs. Curr. Opin. Toxicol. 2018, 10, 46–53. [Google Scholar] [CrossRef]
- Uetrecht, J. Mechanisms of idiosyncratic drug-induced liver injury. Adv. Pharmacol. 2019, 85, 133–163. [Google Scholar] [CrossRef]
- Katarey, D.; Verma, S. Drug-induced liver injury. Clin. Med. 2016, 16, s104–s109. [Google Scholar] [CrossRef]
- Ali, S.E.; Waddington, J.C.; Park, B.K.; Meng, X. Definition of the Chemical and Immunological Signals Involved in Drug-Induced Liver Injury. Chem. Res. Toxicol. 2020, 33, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Mossanen, J.C.; Tacke, F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 2014, 3, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Lefebvre, A.T.; Horuzsko, A. Kupffer Cell Metabolism and Function. J. Enzymol. Metab. 2015, 1, 101. [Google Scholar] [PubMed]
- Slevin, E.; Baiocchi, L.; Wu, N.; Ekser, B.; Sato, K.; Lin, E.; Ceci, L.; Chen, L.; Lorenzo, S.R.; Xu, W.; et al. Kupffer Cells: Inflammation Pathways and Cell-Cell Interactions in Alcohol-Associated Liver Disease. Am. J. Pathol. 2020, 190, 2185–2193. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Ukairo, O.; Khetani, S.R.; McVay, M.; Kanchagar, C.; Seghezzi, W.; Ayanoglu, G.; Irrechukwu, O.; Evers, R. Establishment of a hepatocyte-kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab. Dispos. 2015, 43, 774–785. [Google Scholar] [CrossRef]
- Ogese, M.O.; Faulkner, L.; Jenkins, R.E.; French, N.S.; Copple, I.M.; Antoine, D.J.; Elmasry, M.; Malik, H.; Goldring, C.E.; Park, B.K.; et al. Characterization of Drug-Specific Signaling between Primary Human Hepatocytes and Immune Cells. Toxicol. Sci. 2017, 158, 76–89. [Google Scholar] [CrossRef]
- Naisbitt, D.J.; Olsson-Brown, A.; Gibson, A.; Meng, X.; Ogese, M.O.; Tailor, A.; Thomson, P. Immune dysregulation increases the incidence of delayed-type drug hypersensitivity reactions. Allergy 2020, 75, 781–797. [Google Scholar] [CrossRef]
- Agnelli, G.; Eriksson, B.I.; Cohen, A.T.; Bergqvist, D.; Dahl, O.E.; Lassen, M.R.; Mouret, P.; Rosencher, N.; Andersson, M.; Bylock, A.; et al. Safety assessment of new antithrombotic agents: Lessons from the EXTEND study on ximelagatran. Thromb. Res. 2009, 123, 488–497. [Google Scholar] [CrossRef]
- Singer, J.B.; Lewitzky, S.; Leroy, E.; Yang, F.; Zhao, X.; Klickstein, L.; Wright, T.M.; Meyer, J.; Paulding, C.A. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat. Genet. 2010, 42, 711–714. [Google Scholar] [CrossRef]
- Chitturi, S.; Farrell, G.C. Identifying who is at risk of drug-induced liver injury: Is human leukocyte antigen specificity the key? Hepatology 2011, 53, 358–362. [Google Scholar] [CrossRef]
- Tettey, J.N.; Maggs, J.L.; Rapeport, W.G.; Pirmohamed, M.; Park, B.K. Enzyme-induction dependent bioactivation of troglitazone and troglitazone quinone in vivo. Chem. Res. Toxicol. 2001, 14, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Wu, C.Q.; Li, Z.J.; Liu, Y.; Fan, X.; Wang, Q.J.; Ding, R.G. Characterizing the mechanism of thiazolidinedione-induced hepatotoxicity: An in vitro model in mitochondria. Toxicol. Appl. Pharmacol. 2015, 284, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.E.; Trauner, M.; van Staden, C.J.; Lee, P.H.; Ramachandran, B.; Eschenberg, M.; Afshari, C.A.; Qualls, C.W., Jr.; Lightfoot-Dunn, R.; Hamadeh, H.K. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol. Sci. 2010, 118, 485–500. [Google Scholar] [CrossRef]
- Sidenius, U.; Skonberg, C.; Olsen, J.; Hansen, S.H. In vitro reactivity of carboxylic acid-CoA thioesters with glutathione. Chem. Res. Toxicol. 2004, 17, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Skonberg, C.; Olsen, J.; Madsen, K.G.; Hansen, S.H.; Grillo, M.P. Metabolic activation of carboxylic acids. Expert Opin. Drug Metab. Toxicol. 2008, 4, 425–438. [Google Scholar] [CrossRef]
- Yang, H.; Park, T.; Park, D.; Kang, M.G. Trovafloxacin drives inflammation-associated drug-induced adverse hepatic reaction by changing macrophage polarization. Toxicol. Vitr. 2022, 82, 105374. [Google Scholar] [CrossRef]
- Haasio, K.; Koponen, A.; Penttila, K.E.; Nissinen, E. Effects of entacapone and tolcapone on mitochondrial membrane potential. Eur. J. Pharmacol. 2002, 453, 21–26. [Google Scholar] [CrossRef]
- Smith, K.S.; Smith, P.L.; Heady, T.N.; Trugman, J.M.; Harman, W.D.; Macdonald, T.L. In vitro metabolism of tolcapone to reactive intermediates: Relevance to tolcapone liver toxicity. Chem. Res. Toxicol. 2003, 16, 123–128. [Google Scholar] [CrossRef]
- Kostrubsky, S.E.; Strom, S.C.; Kalgutkar, A.S.; Kulkarni, S.; Atherton, J.; Mireles, R.; Feng, B.; Kubik, R.; Hanson, J.; Urda, E.; et al. Inhibition of hepatobiliary transport as a predictive method for clinical hepatotoxicity of nefazodone. Toxicol. Sci. 2006, 90, 451–459. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Vaz, A.D.; Lame, M.E.; Henne, K.R.; Soglia, J.; Zhao, S.X.; Abramov, Y.A.; Lombardo, F.; Collin, C.; Hendsch, Z.S.; et al. Bioactivation of the nontricyclic antidepressant nefazodone to a reactive quinone-imine species in human liver microsomes and recombinant cytochrome P450 3A4. Drug Metab. Dispos. 2005, 33, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.T.; Kirkham, N.; Johnson, M.K.; Lordan, J.L.; Fisher, A.J.; Peacock, A.J. Sitaxentan-related acute liver failure in a patient with pulmonary arterial hypertension. Eur. Respir. J. 2011, 37, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Maggs, J.L.; Antoine, D.J.; Williams, D.P.; Smith, D.A.; Park, B.K. Role of reactive metabolites in drug-induced hepatotoxicity. Handb. Exp. Pharmacol. 2010, 196, 165–194. [Google Scholar] [CrossRef]
- Faulkner, L.; Meng, X.; Park, B.K.; Naisbitt, D.J. The importance of hapten-protein complex formation in the development of drug allergy. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 293–300. [Google Scholar] [CrossRef]
- Uetrecht, J.P. Is it possible to more accurately predict which drug candidates will cause idiosyncratic drug reactions? Curr. Drug Metab. 2000, 1, 133–141. [Google Scholar] [CrossRef]
- Christ, D.D.; Satoh, H.; Kenna, J.G.; Pohl, L.R. Potential metabolic basis for enflurane hepatitis and the apparent cross-sensitization between enflurane and halothane. Drug Metab. Dispos. 1988, 1, 135–140. [Google Scholar]
- Inman, W.H.; Mushin, W.W. Jaundice after repeated exposure to halothane: An analysis of Reports to the Committee on Safety of Medicines. Br. Med. J. 1974, 1, 5–10. [Google Scholar] [CrossRef]
- Jazaeri, F.; Sheibani, M.; Nezamoleslami, S.; Moezi, L.; Dehpour, A.R. Current Models for Predicting Drug-induced Cholestasis: The Role of Hepatobiliary Transport System. Iran. J. Pharm. Res. 2021, 20, 1–21. [Google Scholar] [CrossRef]
- Copple, B.L.; Jaeschke, H.; Klaassen, C.D. Oxidative stress and the pathogenesis of cholestasis. Semin. Liver Dis. 2010, 30, 195–204. [Google Scholar] [CrossRef]
- Gujral, J.S.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology 2003, 38, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Laverty, H.; Srivastava, A.; Antoine, D.J.; Naisbitt, D.; Williams, D.P. Drug bioactivation and protein adduct formation in the pathogenesis of drug-induced toxicity. Chem. Biol. Interact. 2011, 192, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.R.; Uetrecht, J.; Shear, N.H. Idiosyncratic drug reactions: The reactive metabolite syndromes. Lancet 2000, 356, 1587–1591. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Kalgutkar, A.S.; Soglia, J.R.; Zhao, S.X. Can in vitro metabolism-dependent covalent binding data in liver microsomes distinguish hepatotoxic from nonhepatotoxic drugs? An analysis of 18 drugs with consideration of intrinsic clearance and daily dose. Chem. Res. Toxicol. 2008, 21, 1814–1822. [Google Scholar] [CrossRef]
- Evans, D.C.; Watt, A.P.; Nicoll-Griffith, D.A.; Baillie, T.A. Drug-protein adducts: An industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem. Res. Toxicol. 2004, 17, 3–16. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Lim, P.L. Mitochondrial abnormalities—A link to idiosyncratic drug hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 220, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef]
- Han, D.; Dara, L.; Win, S.; Than, T.A.; Yuan, L.; Abbasi, S.Q.; Liu, Z.X.; Kaplowitz, N. Regulation of drug-induced liver injury by signal transduction pathways: Critical role of mitochondria. Trends Pharmacol. Sci. 2013, 34, 243–253. [Google Scholar] [CrossRef]
- Aleo, M.D.; Luo, Y.; Swiss, R.; Bonin, P.D.; Potter, D.M.; Will, Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014, 60, 1015–1022. [Google Scholar] [CrossRef]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef]
- Labbe, G.; Pessayre, D.; Fromenty, B. Drug-induced liver injury through mitochondrial dysfunction: Mechanisms and detection during preclinical safety studies. Fundam. Clin. Pharmacol. 2008, 22, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, J.; Jorga, K.; Zurcher, G.; Schmitt, M.; Sedek, G.; Da Prada, M.; Van Brummelen, P. Pharmacokinetic-pharmacodynamic interaction between the COMT inhibitor tolcapone and single-dose levodopa. Br. J. Clin. Pharmacol. 1995, 40, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Porceddu, M.; Buron, N.; Roussel, C.; Labbe, G.; Fromenty, B.; Borgne-Sanchez, A. Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol. Sci. 2012, 129, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Pauli-Magnus, C.; Meier, P.J. Hepatobiliary transporters and drug-induced cholestasis. Hepatology 2006, 44, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.; Benet, L.Z. Measures of BSEP Inhibition In Vitro Are Not Useful Predictors of DILI. Toxicol. Sci. 2018, 162, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.; Pantze, M.; Jehle, L.; Ponelle, C.; Scheuermann, G.; Lazendic, M.; Gasser, R. Troglitazone-induced intrahepatic cholestasis by an interference with the hepatobiliary export of bile acids in male and female rats. Correlation with the gender difference in troglitazone sulfate formation and the inhibition of the canalicular bile salt export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology 2001, 167, 83–98. [Google Scholar] [CrossRef]
- Garzel, B.; Hu, T.; Li, L.; Lu, Y.; Heyward, S.; Polli, J.; Zhang, L.; Huang, S.M.; Raufman, J.P.; Wang, H. Metformin Disrupts Bile Acid Efflux by Repressing Bile Salt Export Pump Expression. Pharm. Res. 2020, 37, 26. [Google Scholar] [CrossRef]
- Garzel, B.; Zhang, L.; Huang, S.M.; Wang, H. A Change in Bile Flow: Looking Beyond Transporter Inhibition in the Development of Drug-induced Cholestasis. Curr. Drug Metab. 2019, 20, 621–632. [Google Scholar] [CrossRef]
- Garzel, B.; Yang, H.; Zhang, L.; Huang, S.M.; Polli, J.E.; Wang, H. The role of bile salt export pump gene repression in drug-induced cholestatic liver toxicity. Drug Metab. Dispos. 2014, 42, 318–322. [Google Scholar] [CrossRef]
- Yang, K.; Woodhead, J.L.; Watkins, P.B.; Howell, B.A.; Brouwer, K.L. Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin. Pharmacol. Ther. 2014, 96, 589–598. [Google Scholar] [CrossRef]
- Kock, K.; Ferslew, B.C.; Netterberg, I.; Yang, K.; Urban, T.J.; Swaan, P.W.; Stewart, P.W.; Brouwer, K.L. Risk factors for development of cholestatic drug-induced liver injury: Inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab. Dispos. 2014, 42, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Jee, A.; Sernoskie, S.C.; Uetrecht, J. Idiosyncratic Drug-Induced Liver Injury: Mechanistic and Clinical Challenges. Int. J. Mol. Sci. 2021, 22, 2954. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Pessayre, D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, S.; Vermeulen, N.P.; Gerets, H.H.; Hewitt, P.G.; Ingelman-Sundberg, M.; Park, B.K.; Juhila, S.; Snoeys, J.; Weaver, R.J. Evidence-based selection of training compounds for use in the mechanism-based integrated prediction of drug-induced liver injury in man. Arch. Toxicol. 2016, 90, 2979–3003. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2019, 401, 31–46. [Google Scholar] [CrossRef]
- Pirovino, M.; Muller, O.; Zysset, T.; Honegger, U. Amiodarone-induced hepatic phospholipidosis: Correlation of morphological and biochemical findings in an animal model. Hepatology 1988, 8, 591–598. [Google Scholar] [CrossRef]
- Li, J.G.; Yang, T.C.; Yu, D.M.; Ren, T.H. Fatal acute liver failure after intravenous amiodarone administration. J. Formos. Med. Assoc. 2015, 114, 294–296. [Google Scholar] [CrossRef]
- Shenton, J.M.; Chen, J.; Uetrecht, J.P. Animal models of idiosyncratic drug reactions. Chem. Biol. Interact. 2004, 150, 53–70. [Google Scholar] [CrossRef]
- Kuna, L.; Bozic, I.; Kizivat, T.; Bojanic, K.; Mrso, M.; Kralj, E.; Smolic, R.; Wu, G.Y.; Smolic, M. Models of Drug Induced Liver Injury (DILI)—Current Issues and Future Perspectives. Curr. Drug Metab. 2018, 19, 830–838. [Google Scholar] [CrossRef]
- Lucena, M.I.; Andrade, R.J.; Gomez-Outes, A.; Rubio, M.; Cabello, M.R. Acute liver failure after treatment with nefazodone. Dig. Dis. Sci. 1999, 44, 2577–2579. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; Panel Members; EASL Governing Board. EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 2019, 70, 1222–1261. [Google Scholar] [CrossRef] [PubMed]
- Melino, M.; Gadd, V.L.; Walker, G.V.; Skoien, R.; Barrie, H.D.; Jothimani, D.; Horsfall, L.; Jones, A.; Sweet, M.J.; Thomas, G.P.; et al. Macrophage secretory products induce an inflammatory phenotype in hepatocytes. World J. Gastroenterol. 2012, 18, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, B.D.; King, B.M.; Hasan, M.A.; Alexopoulos, L.G.; Farazi, P.A.; Hendriks, B.S.; Griffith, L.G.; Sorger, P.K.; Tidor, B.; Xu, J.J.; et al. Synergistic drug-cytokine induction of hepatocellular death as an in vitro approach for the study of inflammation-associated idiosyncratic drug hepatotoxicity. Toxicol. Appl. Pharmacol. 2009, 237, 317–330. [Google Scholar] [CrossRef]
- Kegel, V.; Pfeiffer, E.; Burkhardt, B.; Liu, J.L.; Zeilinger, K.; Nussler, A.K.; Seehofer, D.; Damm, G. Subtoxic Concentrations of Hepatotoxic Drugs Lead to Kupffer Cell Activation in a Human In Vitro Liver Model: An Approach to Study DILI. Mediat. Inflamm. 2015, 2015, 640631. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Deguchi, J.; Nishio, N.; Nomura, N.; Funabashi, H. Hepatotoxicants induce cytokine imbalance in response to innate immune system. J. Toxicol. Sci. 2015, 40, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Lerche-Langrand, C.; Toutain, H.J. Precision-cut liver slices: Characteristics and use for in vitro pharmaco-toxicology. Toxicology 2000, 153, 221–253. [Google Scholar] [CrossRef] [PubMed]
- Vernetti, L.A.; Vogt, A.; Gough, A.; Taylor, D.L. Evolution of Experimental Models of the Liver to Predict Human Drug Hepatotoxicity and Efficacy. Clin. Liver Dis. 2017, 21, 197–214. [Google Scholar] [CrossRef]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knospel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef]
- Peng, L.; Zhong, X. Epigenetic regulation of drug metabolism and transport. Acta Pharm. Sin. B 2015, 5, 106–112. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Pioronska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wolfl, S.; Meyer, C.; et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J. Exp. Clin. Cancer Res. 2018, 37, 211. [Google Scholar] [CrossRef]
- Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Negro, A.; Herrera, G.; Castell, J.V.; O’Connor, J.E.; Gomez-Lechon, M.J. Cytometric analysis for drug-induced steatosis in HepG2 cells. Chem. Biol. Interact. 2009, 181, 417–423. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, D.F.; Soars, M.G.; Urbanowicz, R.A.; Riley, R.J. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab. Dispos. 2004, 32, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Sison-Young, R.L.; Lauschke, V.M.; Johann, E.; Alexandre, E.; Antherieu, S.; Aerts, H.; Gerets, H.H.J.; Labbe, G.; Hoet, D.; Dorau, M.; et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Arch. Toxicol. 2017, 91, 1385–1400. [Google Scholar] [CrossRef] [PubMed]
- Underhill, G.H.; Khetani, S.R. Bioengineered Liver Models for Drug Testing and Cell Differentiation Studies. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 426–439.e1. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, M.; He, Z.; Wei, Q.; Li, J. The Biological Function of Kupffer Cells in Liver Disease. In Biology of Myelomonocytic Cells; Books on Demand: Norderstedt, Germany, 2017; Volume 2. [Google Scholar] [CrossRef]
- Kermanizadeh, A.; Brown, D.M.; Moritz, W.; Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Sci. Rep. 2019, 9, 7295. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Kupffer Cells, 3rd ed.; Textbook of Hepatology: From Basic to Clinical Practice; Blackwell Publishing: Ames, IA, USA, 2007. [Google Scholar]
- Chen, W.L.K.; Edington, C.; Suter, E.; Yu, J.; Velazquez, J.J.; Velazquez, J.G.; Shockley, M.; Large, E.M.; Venkataramanan, R.; Hughes, D.J.; et al. Integrated gut/liver microphysiological systems elucidates inflammatory inter-tissue crosstalk. Biotechnol. Bioeng. 2017, 114, 2648–2659. [Google Scholar] [CrossRef]
- Sarkar, U.; Ravindra, K.C.; Large, E.; Young, C.L.; Rivera-Burgos, D.; Yu, J.; Cirit, M.; Hughes, D.J.; Wishnok, J.S.; Lauffenburger, D.A.; et al. Integrated Assessment of Diclofenac Biotransformation, Pharmacokinetics, and Omics-Based Toxicity in a Three-Dimensional Human Liver-Immunocompetent Coculture System. Drug Metab. Dispos. 2017, 45, 855–866. [Google Scholar] [CrossRef]
- Granitzny, A.; Knebel, J.; Muller, M.; Braun, A.; Steinberg, P.; Dasenbrock, C.; Hansen, T. Evaluation of a human in vitro hepatocyte-NPC co-culture model for the prediction of idiosyncratic drug-induced liver injury: A pilot study. Toxicol. Rep. 2017, 4, 89–103. [Google Scholar] [CrossRef]
- Wewering, F.; Jouy, F.; Wissenbach, D.K.; Gebauer, S.; Bluher, M.; Gebhardt, R.; Pirow, R.; von Bergen, M.; Kalkhof, S.; Luch, A.; et al. Characterization of chemical-induced sterile inflammation in vitro: Application of the model compound ketoconazole in a human hepatic co-culture system. Arch. Toxicol. 2017, 91, 799–810. [Google Scholar] [CrossRef]
- Keemink, J.; Oorts, M.; Annaert, P. Primary Hepatocytes in Sandwich Culture. Methods Mol. Biol. 2015, 1250, 175–188. [Google Scholar] [CrossRef]
- Rose, K.A.; Holman, N.S.; Green, A.M.; Andersen, M.E.; LeCluyse, E.L. Co-culture of Hepatocytes and Kupffer Cells as an In Vitro Model of Inflammation and Drug-Induced Hepatotoxicity. J. Pharm. Sci. 2016, 105, 950–964. [Google Scholar] [CrossRef]
- Cui, J.H.; Park, K.; Park, S.R.; Min, B.H. Effects of low-intensity ultrasound on chondrogenic differentiation of mesenchymal stem cells embedded in polyglycolic acid: An in vivo study. Tissue Eng. 2006, 12, 75–82. [Google Scholar] [CrossRef]
- Zinchenko, Y.S.; Schrum, L.W.; Clemens, M.; Coger, R.N. Hepatocyte and kupffer cells co-cultured on micropatterned surfaces to optimize hepatocyte function. Tissue Eng. 2006, 12, 751–761. [Google Scholar] [CrossRef]
- Kostadinova, R.; Boess, F.; Applegate, D.; Suter, L.; Weiser, T.; Singer, T.; Naughton, B.; Roth, A. A long-term three dimensional liver co-culture system for improved prediction of clinically relevant drug-induced hepatotoxicity. Toxicol. Appl. Pharmacol. 2013, 268, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De Toro, J.; Herschlik, L.; Waldner, C.; Mongini, C. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Front. Immunol. 2015, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Du, Y.; Meng, G.; Soon, Y.L.; Sun, S.; Song, N.; Zhang, X.; Xiao, Y.; Wang, J.; Yi, Z.; et al. Long-term functional maintenance of primary human hepatocytes in vitro. Science 2019, 364, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Dallas, S.; Dickmann, L.J.; Fahmi, O.A.; Kenny, J.R.; Kraynov, E.; Nguyen, T.; Patel, A.H.; Slatter, J.G.; Zhang, L. Critical review of preclinical approaches to investigate cytochrome p450-mediated therapeutic protein drug-drug interactions and recommendations for best practices: A white paper. Drug Metab. Dispos. 2013, 41, 1598–1609. [Google Scholar] [CrossRef]
- Corsini, A.; Ganey, P.; Ju, C.; Kaplowitz, N.; Pessayre, D.; Roth, R.; Watkins, P.B.; Albassam, M.; Liu, B.; Stancic, S.; et al. Current challenges and controversies in drug-induced liver injury. Drug Saf. 2012, 35, 1099–1117. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.J.; Liu, S.; Su, G.L.; Vodovotz, Y.; Billiar, T.R. Hepatocytes enhance effects of lipopolysaccharide on liver nonparenchymal cells through close cell interactions. Shock 2005, 23, 453–458. [Google Scholar] [CrossRef]
- Domansky, K.; Inman, W.; Serdy, J.; Dash, A.; Lim, M.H.; Griffith, L.G. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 2010, 10, 51–58. [Google Scholar] [CrossRef]
- Riccalton-Banks, L.; Bhandari, R.; Fry, J.; Shakesheff, K.M. A simple method for the simultaneous isolation of stellate cells and hepatocytes from rat liver tissue. Mol. Cell Biochem. 2003, 248, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.J.; Bhandari, R.; Barrett, D.A.; Bennett, A.J.; Fry, J.R.; Powe, D.; Thomson, B.J.; Shakesheff, K.M. The effect of three-dimensional co-culture of hepatocytes and hepatic stellate cells on key hepatocyte functions in vitro. Cells Tissues Organs. 2005, 181, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.S.; Golberg, I.; Jindal, R.; McCarty, W.J.; Luitje, M.; Hegde, M.; Bhushan, A.; Usta, O.B.; Yarmush, M.L. Long-term coculture strategies for primary hepatocytes and liver sinusoidal endothelial cells. Tissue Eng. Methods 2015, 21, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Rajagopalan, P. 3D hepatic cultures simultaneously maintain primary hepatocyte and liver sinusoidal endothelial cell phenotypes. PLoS ONE 2010, 5, e15456. [Google Scholar] [CrossRef]
- Bale, S.S.; Geerts, S.; Jindal, R.; Yarmush, M.L. Isolation and co-culture of rat parenchymal and non-parenchymal liver cells to evaluate cellular interactions and response. Sci. Rep. 2016, 6, 25329. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Balis, U.J.; Yarmush, M.L.; Toner, M. Microfabrication of hepatocyte/fibroblast co-cultures: Role of homotypic cell interactions. Biotechnol. Prog. 1998, 14, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Balis, U.J.; Yarmush, M.L.; Toner, M. Probing heterotypic cell interactions: Hepatocyte function in microfabricated co-cultures. J. Biomater. Sci. Polym. Ed. 1998, 9, 1137–1160. [Google Scholar] [CrossRef]
- Khetani, S.R.; Kanchagar, C.; Ukairo, O.; Krzyzewski, S.; Moore, A.; Shi, J.; Aoyama, S.; Aleo, M.; Will, Y. Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol. Sci. 2013, 132, 107–117. [Google Scholar] [CrossRef]
- Manning, F.J.; Swartz, M. (Eds.) Review of the Fialuridine (FIAU) Clinical Trials; National Academies Press: Washington, DC, USA, 1995. [Google Scholar]
- Davidson, M.D.; Kukla, D.A.; Khetani, S.R. Microengineered cultures containing human hepatic stellate cells and hepatocytes for drug development. Integr. Biol. 2017, 9, 662–677. [Google Scholar] [CrossRef]
- Edling, Y.; Sivertsson, L.K.; Butura, A.; Ingelman-Sundberg, M.; Ek, M. Increased sensitivity for troglitazone-induced cytotoxicity using a human in vitro co-culture model. Toxicol. Vitr. 2009, 23, 1387–1395. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.; Moro, S.M.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed]
- Mandenius, C.F.; Andersson, T.B.; Alves, P.M.; Batzl-Hartmann, C.; Bjorquist, P.; Carrondo, M.J.; Chesne, C.; Coecke, S.; Edsbagge, J.; Fredriksson, J.M.; et al. Toward preclinical predictive drug testing for metabolism and hepatotoxicity by using in vitro models derived from human embryonic stem cells and human cell lines—A report on the Vitrocellomics EU-project. Altern. Lab. Anim. 2011, 39, 147–171. [Google Scholar] [CrossRef] [PubMed]
- Vivares, A.; Salle-Lefort, S.; Arabeyre-Fabre, C.; Ngo, R.; Penarier, G.; Bremond, M.; Moliner, P.; Gallas, J.F.; Fabre, G.; Klieber, S. Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica 2015, 45, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brown, P.C.; Chow, E.C.Y.; Ewart, L.; Ferguson, S.S.; Fitzpatrick, S.; Freedman, B.S.; Guo, G.L.; Hedrich, W.; Heyward, S.; et al. 3D cell culture models: Drug pharmacokinetics, safety assessment, and regulatory consideration. Clin. Transl. Sci. 2021, 14, 1659–1680. [Google Scholar] [CrossRef]
- Hendriks, D.F.; Fredriksson Puigvert, L.; Messner, S.; Mortiz, W.; Ingelman-Sundberg, M. Hepatic 3D spheroid models for the detection and study of compounds with cholestatic liability. Sci. Rep. 2016, 6, 35434. [Google Scholar] [CrossRef]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, Functional, and Mechanistic Comparisons of Stem Cell-Derived Hepatocytes, HepaRG Cells, and Three-Dimensional Human Hepatocyte Spheroids as Predictive In Vitro Systems for Drug-Induced Liver Injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef]
- Messner, S.; Fredriksson, L.; Lauschke, V.M.; Roessger, K.; Escher, C.; Bober, M.; Kelm, J.M.; Ingelman-Sundberg, M.; Moritz, W. Transcriptomic, Proteomic, and Functional Long-Term Characterization of Multicellular Three-Dimensional Human Liver Microtissues. Appl. In Vitro Toxicol. 2018, 4, 1–12. [Google Scholar] [CrossRef]
- Gupta, R.; Schrooders, Y.; Hauser, D.; van Herwijnen, M.; Albrecht, W.; Ter Braak, B.; Brecklinghaus, T.; Castell, J.V.; Elenschneider, L.; Escher, S.; et al. Comparing in vitro human liver models to in vivo human liver using RNA-Seq. Arch. Toxicol. 2021, 95, 573–589. [Google Scholar] [CrossRef]
- Ardalani, H.; Sengupta, S.; Harms, V.; Vickerman, V.; Thomson, J.A.; Murphy, W.L. 3-D culture and endothelial cells improve maturity of human pluripotent stem cell-derived hepatocytes. Acta Biomater. 2019, 95, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Aihara, E.; Watson, C.; Mourya, R.; Mizuochi, T.; Shivakumar, P.; Phelan, K.; Mayhew, C.; Helmrath, M.; Takebe, T.; et al. Paracrine signals regulate human liver organoid maturation from induced pluripotent stem cells. Development 2017, 144, 1056–1064. [Google Scholar] [CrossRef]
- Kryou, C.; Leva, V.; Chatzipetrou, M.; Zergioti, I. Bioprinting for Liver Transplantation. Bioengineering 2019, 6, 95. [Google Scholar] [CrossRef]
- Messner, S.; Agarkova, I.; Moritz, W.; Kelm, J.M. Multi-cell type human liver microtissues for hepatotoxicity testing. Arch. Toxicol. 2013, 87, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kim, M.H.; Shirahama, H.; Lee, J.H.; Ng, S.S.; Glenn, J.S.; Cho, N.J. ECM proteins in a microporous scaffold influence hepatocyte morphology, function, and gene expression. Sci. Rep. 2016, 6, 37427. [Google Scholar] [CrossRef] [PubMed]
- Knight, E.; Murray, B.; Carnachan, R.; Przyborski, S. Alvetex(R): Polystyrene scaffold technology for routine three dimensional cell culture. Methods Mol. Biol. 2011, 695, 323–340. [Google Scholar] [CrossRef]
- Skardal, A.; Smith, L.; Bharadwaj, S.; Atala, A.; Soker, S.; Zhang, Y. Tissue specific synthetic ECM hydrogels for 3-D in vitro maintenance of hepatocyte function. Biomaterials 2012, 33, 4565–4575. [Google Scholar] [CrossRef]
- Mazza, G.; Rombouts, K.; Rennie Hall, A.; Urbani, L.; Vinh Luong, T.; Al-Akkad, W.; Longato, L.; Brown, D.; Maghsoudlou, P.; Dhillon, A.P.; et al. Decellularized human liver as a natural 3D-scaffold for liver bioengineering and transplantation. Sci. Rep. 2015, 5, 13079. [Google Scholar] [CrossRef]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of Hepatic 2D Sandwich Cultures and 3D Spheroids for Long-term Toxicity Applications: A Multicenter Study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef]
- Proctor, W.R.; Foster, A.J.; Vogt, J.; Summers, C.; Middleton, B.; Pilling, M.A.; Shienson, D.; Kijanska, M.; Strobel, S.; Kelm, J.M.; et al. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch. Toxicol. 2017, 91, 2849–2863. [Google Scholar] [CrossRef] [PubMed]
- Hafiz, E.O.A.; Bulutoglu, B.; Mansy, S.S.; Chen, Y.; Abu-Taleb, H.; Soliman, S.A.M.; El-Hindawi, A.A.F.; Yarmush, M.L.; Uygun, B.E. Development of liver microtissues with functional biliary ductular network. Biotechnol. Bioeng. 2021, 118, 17–29. [Google Scholar] [CrossRef]
- Li, F.; Cao, L.; Parikh, S.; Zuo, R. Three-Dimensional Spheroids with Primary Human Liver Cells and Differential Roles of Kupffer Cells in Drug-Induced Liver Injury. J. Pharm. Sci. 2020, 109, 1912–1923. [Google Scholar] [CrossRef]
- Jalan-Sakrikar, N.; Brevini, T.; Huebert, R.C.; Sampaziotis, F. Organoids and regenerative hepatology. Hepatology 2023, 77, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Hannan, N.R.; Segeritz, C.P.; Touboul, T.; Vallier, L. Production of hepatocyte-like cells from human pluripotent stem cells. Nat. Protoc. 2013, 8, 430–437. [Google Scholar] [CrossRef]
- Holmgren, G.; Sjogren, A.K.; Barragan, I.; Sabirsh, A.; Sartipy, P.; Synnergren, J.; Bjorquist, P.; Ingelman-Sundberg, M.; Andersson, T.B.; Edsbagge, J. Long-term chronic toxicity testing using human pluripotent stem cell-derived hepatocytes. Drug Metab. Dispos. 2014, 42, 1401–1406. [Google Scholar] [CrossRef]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.R.; Dunn, A.; et al. High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell-Derived Organoids. Gastroenterology 2021, 160, 831–846.e10. [Google Scholar] [CrossRef]
- Koido, M.; Kawakami, E.; Fukumura, J.; Noguchi, Y.; Ohori, M.; Nio, Y.; Nicoletti, P.; Aithal, G.P.; Daly, A.K.; Watkins, P.B.; et al. Polygenic architecture informs potential vulnerability to drug-induced liver injury. Nat. Med. 2020, 26, 1541–1548. [Google Scholar] [CrossRef]
- Zhang, C.J.; Meyer, S.R.; O’Meara, M.J.; Huang, S.; Capeling, M.M.; Ferrer-Torres, D.; Childs, C.J.; Spence, J.R.; Fontana, R.J.; Sexton, J.Z. A human liver organoid screening platform for DILI risk prediction. J. Hepatol. 2023, 78, 998–1006. [Google Scholar] [CrossRef]
- Gudapati, H.; Dey, M.; Ozbolat, I. A comprehensive review on droplet-based bioprinting: Past, present and future. Biomaterials 2016, 102, 20–42. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.W.; Lee, S.J.; Ko, I.K.; Kengla, C.; Yoo, J.J.; Atala, A. A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat. Biotechnol. 2016, 34, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Matsusaki, M.; Sakaue, K.; Kadowaki, K.; Akashi, M. Three-dimensional human tissue chips fabricated by rapid and automatic inkjet cell printing. Adv. Healthc. Mater. 2013, 2, 534–539. [Google Scholar] [CrossRef]
- Nguyen, D.G.; Funk, J.; Robbins, J.B.; Crogan-Grundy, C.; Presnell, S.C.; Singer, T.; Roth, A.B. Bioprinted 3D Primary Liver Tissues Allow Assessment of Organ-Level Response to Clinical Drug Induced Toxicity In Vitro. PLoS ONE 2016, 11, e0158674. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Kang, H.W.; Mead, I.; Bishop, C.; Shupe, T.; Lee, S.J.; Jackson, J.; Yoo, J.; Soker, S.; et al. A hydrogel bioink toolkit for mimicking native tissue biochemical and mechanical properties in bioprinted tissue constructs. Acta Biomater. 2015, 25, 24–34. [Google Scholar] [CrossRef]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Shrike Zhang, Y.; Shin, S.R.; Calzone, G.; et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 2016, 8, 014101. [Google Scholar] [CrossRef]
- Lee, J.W.; Choi, Y.J.; Yong, W.J.; Pati, F.; Shim, J.H.; Kang, K.S.; Kang, I.H.; Park, J.; Cho, D.W. Development of a 3D cell printed construct considering angiogenesis for liver tissue engineering. Biofabrication 2016, 8, 015007. [Google Scholar] [CrossRef]
- Grix, T.; Ruppelt, A.; Thomas, A.; Amler, A.K.; Noichl, B.P.; Lauster, R.; Kloke, L. Bioprinting Perfusion-Enabled Liver Equivalents for Advanced Organ-on-a-Chip Applications. Genes 2018, 9, 176. [Google Scholar] [CrossRef]
- Tostoes, R.M.; Leite, S.B.; Serra, M.; Jensen, J.; Bjorquist, P.; Carrondo, M.J.; Brito, C.; Alves, P.M. Human liver cell spheroids in extended perfusion bioreactor culture for repeated-dose drug testing. Hepatology 2012, 55, 1227–1236. [Google Scholar] [CrossRef]
- Rebelo, S.P.; Costa, R.; Silva, M.M.; Marcelino, P.; Brito, C.; Alves, P.M. Three-dimensional co-culture of human hepatocytes and mesenchymal stem cells: Improved functionality in long-term bioreactor cultures. J. Tissue Eng. Regen. Med. 2017, 11, 2034–2045. [Google Scholar] [CrossRef]
- Wang, Y.; Lee, D.; Zhang, L.; Jeon, H.; Mendoza-Elias, J.E.; Harvat, T.A.; Hassan, S.Z.; Zhou, A.; Eddington, D.T.; Oberholzer, J. Systematic prevention of bubble formation and accumulation for long-term culture of pancreatic islet cells in microfluidic device. Biomed. Microdevices 2012, 14, 419–426. [Google Scholar] [CrossRef]
- Farzaneh, Z.; Abbasalizadeh, S.; Asghari-Vostikolaee, M.H.; Alikhani, M.; Cabral, J.M.S.; Baharvand, H. Dissolved oxygen concentration regulates human hepatic organoid formation from pluripotent stem cells in a fully controlled bioreactor. Biotechnol. Bioeng. 2020, 117, 3739–3756. [Google Scholar] [CrossRef]
- Mueller, D.; Tascher, G.; Muller-Vieira, U.; Knobeloch, D.; Nuessler, A.K.; Zeilinger, K.; Heinzle, E.; Noor, F. In-depth physiological characterization of primary human hepatocytes in a 3D hollow-fiber bioreactor. J. Tissue Eng. Regen. Med. 2011, 5, e207–e218. [Google Scholar] [CrossRef]
- Rowe, C.; Shaeri, M.; Large, E.; Cornforth, T.; Robinson, A.; Kostrzewski, T.; Sison-Young, R.; Goldring, C.; Park, K.; Hughes, D. Perfused human hepatocyte microtissues identify reactive metabolite-forming and mitochondria-perturbing hepatotoxins. Toxicol. Vitr. 2018, 46, 29–38. [Google Scholar] [CrossRef]
- Tostoes, R.M.; Leite, S.B.; Miranda, J.P.; Sousa, M.; Wang, D.I.; Carrondo, M.J.; Alves, P.M. Perfusion of 3D encapsulated hepatocytes—A synergistic effect enhancing long-term functionality in bioreactors. Biotechnol. Bioeng. 2011, 108, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.P.; Shipley, R.; Ellis, M.J.; Webb, S.; Ward, J.; Gardner, I.; Creton, S. Novel in vitro and mathematical models for the prediction of chemical toxicity. Toxicol. Res. 2013, 2, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.C.; Encke, J.; Hole, O.; Muller, C.; Ryan, C.J.; Neuhaus, P. Bioreactor for a larger scale hepatocyte in vitro perfusion. Transplantation 1994, 58, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.C.; Mutig, K.; Sauer, I.M.; Schrade, P.; Efimova, E.; Mieder, T.; Naumann, G.; Grunwald, A.; Pless, G.; Mas, A.; et al. Use of primary human liver cells originating from discarded grafts in a bioreactor for liver support therapy and the prospects of culturing adult liver stem cells in bioreactors: A morphologic study. Transplantation 2003, 76, 781–786. [Google Scholar] [CrossRef]
- Zeilinger, K.; Schreiter, T.; Darnell, M.; Soderdahl, T.; Lubberstedt, M.; Dillner, B.; Knobeloch, D.; Nussler, A.K.; Gerlach, J.C.; Andersson, T.B. Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng. Methods 2011, 17, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Lubberstedt, M.; Muller-Vieira, U.; Biemel, K.M.; Darnell, M.; Hoffmann, S.A.; Knospel, F.; Wonne, E.C.; Knobeloch, D.; Nussler, A.K.; Gerlach, J.C.; et al. Serum-free culture of primary human hepatocytes in a miniaturized hollow-fibre membrane bioreactor for pharmacological in vitro studies. J. Tissue Eng. Regen. Med. 2015, 9, 1017–1026. [Google Scholar] [CrossRef]
- Jang, K.J.; Otieno, M.A.; Ronxhi, J.; Lim, H.K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 2019, 11, eaax5516. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, N.; Yang, H.; Luo, C.; Gong, Y.; Tong, C.; Gao, Y.; Lu, S.; Long, M. Mimicking liver sinusoidal structures and functions using a 3D-configured microfluidic chip. Lab Chip 2017, 17, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.Y.; Seok, J.I.; Kim, D.S. Flow-Based Three-Dimensional Co-Culture Model for Long-Term Hepatotoxicity Prediction. Micromachines 2019, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Rubiano, A.; Indapurkar, A.; Yokosawa, R.; Miedzik, A.; Rosenzweig, B.; Arefin, A.; Moulin, C.M.; Dame, K.; Hartman, N.; Volpe, D.A.; et al. Characterizing the reproducibility in using a liver microphysiological system for assaying drug toxicity, metabolism, and accumulation. Clin. Transl. Sci. 2021, 14, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, U.; Rivera-Burgos, D.; Large, E.M.; Hughes, D.J.; Ravindra, K.C.; Dyer, R.L.; Ebrahimkhani, M.R.; Wishnok, J.S.; Griffith, L.G.; Tannenbaum, S.R. Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab. Dispos. 2015, 43, 1091–1099. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, J.X.; Lauschke, V.M. Comprehensive Evaluation of Organotypic and Microphysiological Liver Models for Prediction of Drug-Induced Liver Injury. Front. Pharmacol. 2019, 10, 1093. [Google Scholar] [CrossRef]
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox. Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef]
- Lee-Montiel, F.T.; George, S.M.; Gough, A.H.; Sharma, A.D.; Wu, J.; DeBiasio, R.; Vernetti, L.A.; Taylor, D.L. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med. 2017, 242, 1617–1632. [Google Scholar] [CrossRef]
- Wang, Y.I.; Carmona, C.; Hickman, J.J.; Shuler, M.L. Multiorgan Microphysiological Systems for Drug Development: Strategies, Advances, and Challenges. Adv. Healthc. Mater. 2018, 7, 1701000. [Google Scholar] [CrossRef]
- Haque, A.; Gheibi, P.; Gao, Y.; Foster, E.; Son, K.J.; You, J.; Stybayeva, G.; Patel, D.; Revzin, A. Cell biology is different in small volumes: Endogenous signals shape phenotype of primary hepatocytes cultured in microfluidic channels. Sci. Rep. 2016, 6, 33980. [Google Scholar] [CrossRef]
- Novik, E.I.; Dwyer, J.; Morelli, J.K.; Parekh, A.; Cho, C.; Pludwinski, E.; Shrirao, A.; Freedman, R.M.; MacDonald, J.S.; Jayyosi, Z. Long-enduring primary hepatocyte-based co-cultures improve prediction of hepatotoxicity. Toxicol. Appl. Pharmacol. 2017, 336, 20–30. [Google Scholar] [CrossRef]
- Ewart, L.; Apostolou, A.; Briggs, S.A.; Carman, C.V.; Chaff, J.T.; Heng, A.R.; Jadalannagari, S.; Janardhanan, J.; Jang, K.J.; Joshipura, S.R.; et al. Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology. Commun. Med. 2022, 2, 154. [Google Scholar] [CrossRef] [PubMed]
- Rennert, K.; Steinborn, S.; Groger, M.; Ungerbock, B.; Jank, A.M.; Ehgartner, J.; Nietzsche, S.; Dinger, J.; Kiehntopf, M.; Funke, H.; et al. A microfluidically perfused three dimensional human liver model. Biomaterials 2015, 71, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ott, L.M.; Ramachandran, K.; Stehno-Bittel, L. An Automated Multiplexed Hepatotoxicity and CYP Induction Assay Using HepaRG Cells in 2D and 3D. SLAS Discov. 2017, 22, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Ramaiahgari, S.C.; Waidyanatha, S.; Dixon, D.; DeVito, M.J.; Paules, R.S.; Ferguson, S.S. From the Cover: Three-Dimensional (3D) HepaRG Spheroid Model with Physiologically Relevant Xenobiotic Metabolism Competence and Hepatocyte Functionality for Liver Toxicity Screening. Toxicol. Sci. 2017, 159, 124–136. [Google Scholar] [CrossRef]
- Takayama, K.; Kawabata, K.; Nagamoto, Y.; Kishimoto, K.; Tashiro, K.; Sakurai, F.; Tachibana, M.; Kanda, K.; Hayakawa, T.; Furue, M.K.; et al. 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials 2013, 34, 1781–1789. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Prediction of Drug-Induced Hepatotoxicity Using Long-Term Stable Primary Hepatic 3D Spheroid Cultures in Chemically Defined Conditions. Toxicol. Sci. 2018, 163, 655–665. [Google Scholar] [CrossRef]
- Ramaiahgari, S.C.; den Braver, M.W.; Herpers, B.; Terpstra, V.; Commandeur, J.N.; van de Water, B.; Price, L.S. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014, 88, 1083–1095. [Google Scholar] [CrossRef]
- Liu, J.; Li, R.; Xue, R.; Li, T.; Leng, L.; Wang, Y.; Wang, J.; Ma, J.; Yan, J.; Yan, F.; et al. Liver Extracellular Matrices Bioactivated Hepatic Spheroids as a Model System for Drug Hepatotoxicity Evaluations. Adv. Biosyst. 2018, 2. [Google Scholar] [CrossRef]
- Ware, B.R.; Berger, D.R.; Khetani, S.R. Prediction of Drug-Induced Liver Injury in Micropatterned Co-cultures Containing iPSC-Derived Human Hepatocytes. Toxicol. Sci. 2015, 145, 252–262. [Google Scholar] [CrossRef]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; van Grunsven, L.A. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, O.; Hancock, M.K.; Hesley, J.; Hong, D.; Cohen, A.; Gentry, J.; Carlson, C.B.; Mann, D.A. Phenotypic Characterization of Toxic Compound Effects on Liver Spheroids Derived from iPSC Using Confocal Imaging and Three-Dimensional Image Analysis. Assay Drug Dev. Technol. 2016, 14, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drugs | Date Approved | Withdrawn Date | Reason for Withdrawal | Potential Mechanism of DILI |
|---|---|---|---|---|
| Troglitazone | 29 January 1997 | 21 March 2000 | Liver failure | GSH depletion [42], reactive species formation [43], and BSEP inhibition [44] |
| Bromfenac | 15 July 1997 | 22 June 1998 | Severe hepatitis and liver failure | GSH depletion and reactive species formation [45,46] |
| Trovafloxacin | 1997 | 15 June 1999 | Acute hepatitis | Immune-mediated [47] |
| Tolcapone | 25 January 1998 & 31 August 2009 | November 1998 | Acute liver failure | Mitochondrial dysfunction and reactive species formation [48,49] |
| Nefazodone | 5 May 2003 | 14 June 2004 | Acute liver failure | BSEP inhibition [50] and reactive species formation [51,52] |
| Sitaxentan | 15 June 2007 | 10 December 2010 | Liver injury | BSEP inhibition [53] |
| Model | Challenges | Advantages | References | |
|---|---|---|---|---|
| 2D | Monoculture + soluble fraction | No cell–cell interaction | Easy to culture, improved hepatic phenotype | [93,94] |
| Mixed culture | Random distribution of cells | Physical cell–cell interaction | [114] | |
| Transwell | Heterogenic cell–cell interaction, degree of separation | Increased expression of pro-inflammatory cytokines/chemokines | [131] | |
| Micropattern | Cell–ECM detachment, no direct heterogenic cell–cell interaction | Phenotypic stability | [112] | |
| 3D | Spheroids & organoids | Potential for toxicity from synthetic scaffolds, increased potential of false positive for drugs | Viable for the long term and maintained stable transcriptomes/proteomes | [157] |
| Bioreactors | Not amenable to high-throughput screening, hydrodynamic shear forces | Concentration nutrient gradient, maintained hepatic metabolic function | [179] | |
| Microfluidic liver-on-a-chip | Lack standardized protocol, not amenable to high throughput | 3D architecture, layers of NPCs adjacent to hepatocytes, extracellular matrix | [190] | |
| Bioprinting | Not amenable to high-throughput screening, expensive | High resolution, 3D architecture, and phenotypic stability | [172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stern, S.; Wang, H.; Sadrieh, N. Microphysiological Models for Mechanistic-Based Prediction of Idiosyncratic DILI. Cells 2023, 12, 1476. https://doi.org/10.3390/cells12111476
Stern S, Wang H, Sadrieh N. Microphysiological Models for Mechanistic-Based Prediction of Idiosyncratic DILI. Cells. 2023; 12(11):1476. https://doi.org/10.3390/cells12111476
Chicago/Turabian StyleStern, Sydney, Hongbing Wang, and Nakissa Sadrieh. 2023. "Microphysiological Models for Mechanistic-Based Prediction of Idiosyncratic DILI" Cells 12, no. 11: 1476. https://doi.org/10.3390/cells12111476
APA StyleStern, S., Wang, H., & Sadrieh, N. (2023). Microphysiological Models for Mechanistic-Based Prediction of Idiosyncratic DILI. Cells, 12(11), 1476. https://doi.org/10.3390/cells12111476