
CAR-Based Therapy for Autoimmune Diseases: A Novel Powerful Option



Abstract
:
1. Introduction
Adoptive Cell Transfer Immunotherapy Approaches
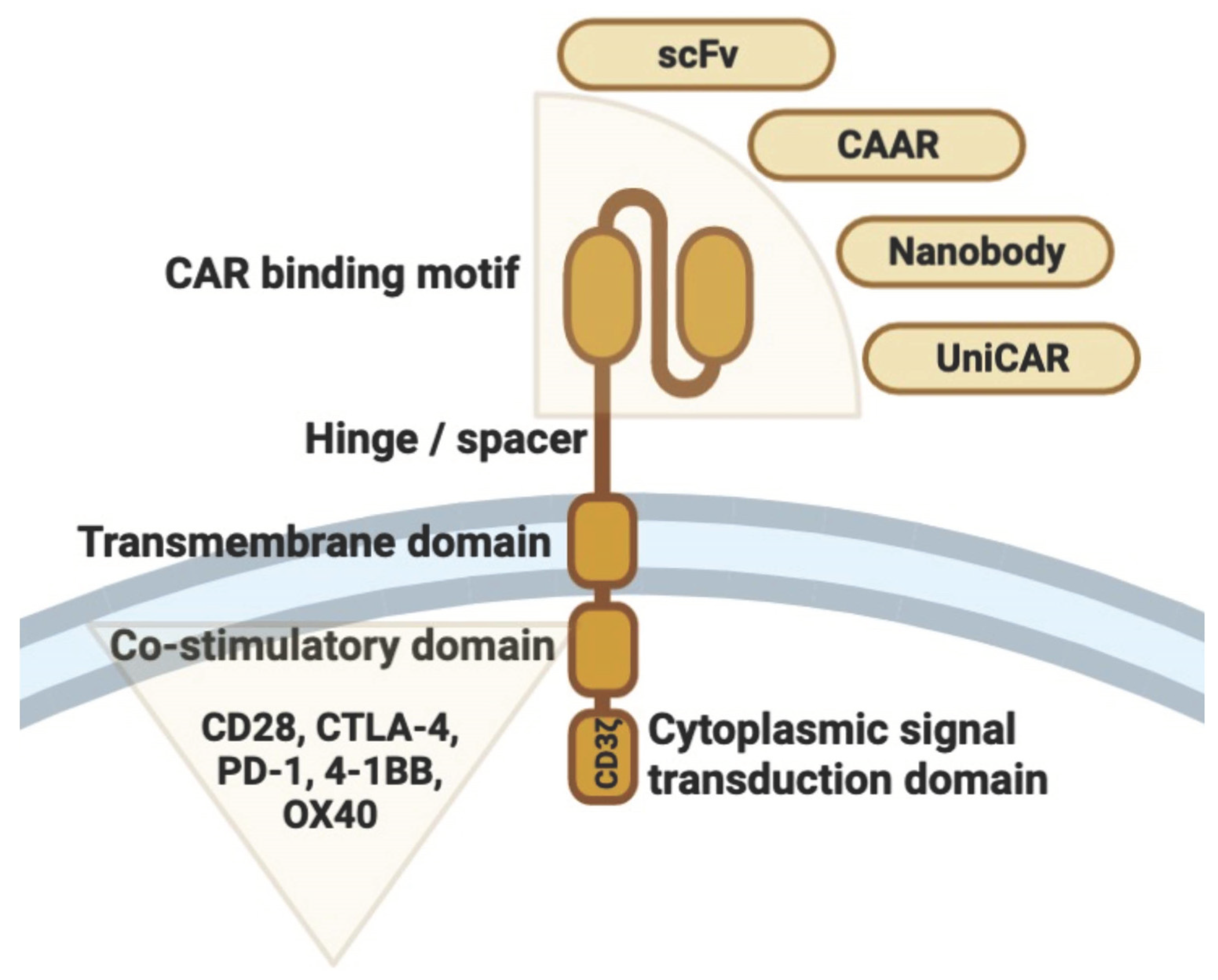
2. Structural Design and T Cell Engineering
2.1. T Cells as the Basis of Engineering
2.2. CAR T Cell Manufacturing
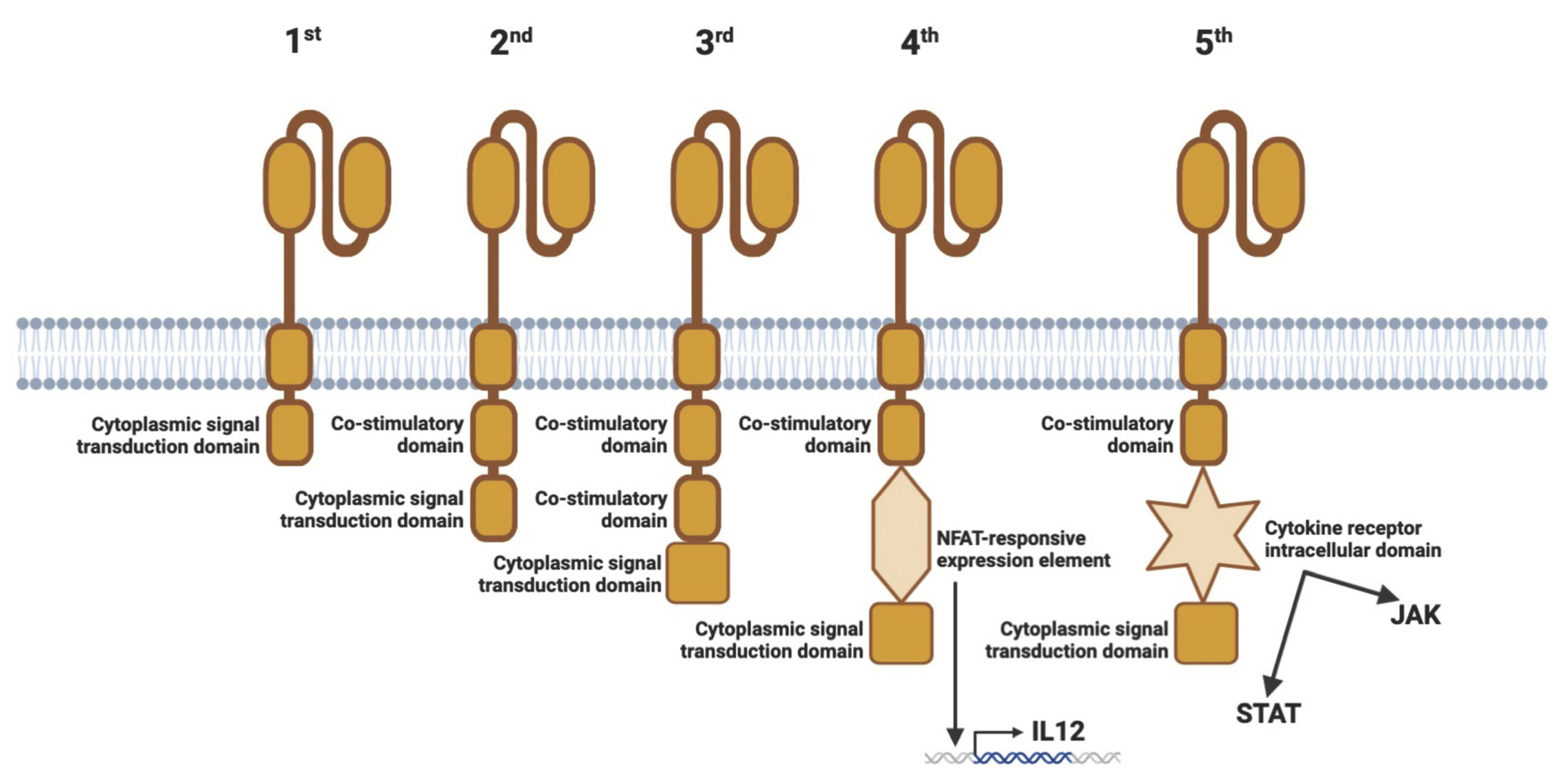
2.3. Generations of CARs
3. CAR-Based Therapy for Autoimmune and Immune-Mediated Diseases
3.1. CAR T and CAR Treg Therapies and Clinical Trials in Systemic Lupus Erythematosus
3.2. CAR T and CAAR NK Therapies and Clinical Trials in Sjögren’s Syndrome
3.3. Compound CAR T Therapy and Clinical Trials in ANCA-Associated Vasculitis and Autoimmune Hemolytic Anemia
3.4. CAR T and CAR Treg Therapies in Rheumatoid Arthritis
3.5. CAR T Therapies and Clinical Trials in Systemic Sclerosis
3.6. CAR T, CAAR T and CAR Treg Therapies and Clinical Trials in Immune-Mediated Neurological Disorders
3.7. CAAR T Therapies and Clinical Trials in Pemphigus Vulgaris
3.8. CAR T and CAR Treg Therapies and Clinical Trials in Dermatomyositis, Adult-Onset Still’s Disease, and Inflammatory Bowel Disease
3.9. CAR T Therapy in Type 1 Diabetes Mellitus
3.10. CAR Treg Therapy and Clinical Trials in Graft-Versus-Host Disease
4. Limitations of CAR-Based Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; van der Waart, A.B. Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytom. Part A 2019, 95, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.E.; Jenkinson, W.E.; Anderson, G. Thymus medulla fosters generation of natural Treg cells, invariant γδ T cells, and invariant NKT cells: What we learn from intrathymic migration. Eur. J. Immunol. 2015, 45, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.; Payne, A.S. Engineering Cell Therapies for Autoimmune Diseases: From Preclinical to Clinical Proof of Concept. Immune Netw. 2022, 22, e37. [Google Scholar] [CrossRef] [PubMed]
- Sanz, I.; Wei, C.; Jenks, S.A.; Cashman, K.S.; Tipton, C.; Woodruff, M.C.; Hom, J.; Lee, F.E.-H. Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations. Front. Immunol. 2019, 10, 2458. [Google Scholar] [CrossRef] [Green Version]
- Lallana, E.C.; Fadul, C.E. Toxicities of Immunosuppressive Treatment of Autoimmune Neurologic Diseases. Curr. Neuropharmacol. 2011, 9, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. Biodrugs 2019, 33, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.M.; Kim, W.-U. Targeted Immunotherapy for Autoimmune Disease. Immune Netw. 2022, 22, e9. [Google Scholar] [CrossRef]
- Rosman, Z.; Shoenfeld, Y.; Zandman-Goddard, G. Biologic therapy for autoimmune diseases: An update. BMC Med. 2013, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Lee, D.S.W.; Rojas, O.L.; Gommerman, J.L. B cell depletion therapies in autoimmune disease: Advances and mechanistic insights. Nat. Rev. Drug Discov. 2020, 20, 179–199. [Google Scholar] [CrossRef]
- Tony, H.P.; Burmester, G.; Schulze-Koops, H.; Grunke, M.; Henes, J.; Kötter, I.; Haas, J.; Unger, L.; Lovric, S.; Haubitz, M.; et al. Safety and clinical outcomes of rituximab therapy in patients with different autoimmune diseases: Experience from a national registry (GRAID). Arthritis Res. Ther. 2011, 13, R75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Spiess, P.; LaFreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 2020, 11, 1689. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. Ebiomedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Baulu, E.; Gardet, C.; Chuvin, N.; Depil, S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci. Adv. 2023, 9, eadf3700. [Google Scholar] [CrossRef]
- Adabi, N.; Pordel, S.; Rezaee, M.A.; Shobeiri, F.S.; Shobeiri, S.S. Application of CAR-T cell technology in autoimmune diseases and human immunodeficiency virus infection treatment. J. Gene Med. 2023, 25, e3484. [Google Scholar] [CrossRef]
- Włodarczyk, M.; Pyrzynska, B. CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer. Cancers 2022, 15, 117. [Google Scholar] [CrossRef]
- Moreno, C.; Haynie, C.; Johnson, A.; Weber, K.S. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines 2022, 10, 1493. [Google Scholar] [CrossRef]
- Boulch, M.; Cazaux, M.; Loe-Mie, Y.; Thibaut, R.; Corre, B.; Lemaître, F.; Grandjean, C.L.; Garcia, Z.; Bousso, P. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci. Immunol. 2021, 6, eabd4344. [Google Scholar] [CrossRef] [PubMed]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. Faculty Opinions recommendation of A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. Faculty Opinions recommendation of A distinct function of regulatory T cells in tissue protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempkes, R.; Joosten, I.; Koenen, H.J.P.M.; He, X. Metabolic Pathways Involved in Regulatory T Cell Functionality. Front. Immunol. 2019, 10, 2839. [Google Scholar] [CrossRef]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef]
- Curotto de Lafaille, M.A.; Lafaille, J.J. Natural and adaptive foxp3+ regulatory T cells: More of the same or a division of labor? Immunity 2009, 30, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.-L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef]
- Barnes, M.J.; Griseri, T.; Johnson, A.M.F.; Young, W.; Powrie, F.; Izcue, A. CTLA-4 promotes Foxp3 induction and regulatory T cell accumulation in the intestinal lamina propria. Mucosal Immunol. 2013, 6, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [Green Version]
- Gravano, D.M.; Vignali, D.A.A. The battle against immunopathology: Infectious tolerance mediated by regulatory T cells. Cell. Mol. Life Sci. 2012, 69, 1997–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juścińska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Młynarski, W.; et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets—Results of one year follow-up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Dall’Era, M.; Pauli, M.L.; Remedios, K.; Taravati, K.; Sandoval, P.M.; Putnam, A.L.; Lares, A.; Haemel, A.; Tang, Q.; Hellerstein, M.; et al. Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 431–440. [Google Scholar] [CrossRef]
- Safety and Efficacy Study of Regulatory T Cells in Treating Autoimmune Hepatitis. Available online: https://ClinicalTrials.gov/show/NCT02704338 (accessed on 12 April 2023).
- Goldberg, R.; Scotta, C.; Cooper, D.; Nissim-Eliraz, E.; Nir, E.; Tasker, S.; Irving, P.M.; Sanderson, J.; Lavender, P.; Ibrahim, F.; et al. Correction of Defective T-Regulatory Cells From Patients With Crohn’s Disease by Ex Vivo Ligation of Retinoic Acid Receptor-α. Gastroenterology 2019, 156, 1775–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skuljec, J.; Chmielewski, M.; Happle, C.; Habener, A.; Busse, M.; Abken, H.; Hansen, G. Chimeric Antigen Receptor-Redirected Regulatory T Cells Suppress Experimental Allergic Airway Inflammation, a Model of Asthma. Front. Immunol. 2017, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Dawson, N.A.J.; Rosado-Sánchez, I.; Novakovsky, G.E.; Fung, V.C.W.; Huang, Q.; McIver, E.; Sun, G.; Gillies, J.; Speck, M.; Orban, P.C.; et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci. Transl. Med. 2020, 12, eaaz3866. [Google Scholar] [CrossRef]
- Yeh, W.-I.; Seay, H.R.; Newby, B.; Posgai, A.L.; Moniz, F.B.; Michels, A.; Mathews, C.E.; Bluestone, J.A.; Brusko, T.M. Avidity and Bystander Suppressive Capacity of Human Regulatory T Cells Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front. Immunol. 2017, 8, 1313. [Google Scholar] [CrossRef]
- Fu, R.Y.; Chen, A.C.; Lyle, M.J.; Chen, C.-Y.; Liu, C.L.; Miao, C.H. CD4+ T cells engineered with FVIII-CAR and murine Foxp3 suppress anti-factor VIII immune responses in hemophilia a mice. Cell. Immunol. 2020, 358, 104216. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, E.; Xie, X.; Wang, J.; Zheng, H.; Ju, Y.; Chen, L.; Li, C.; Zhou, X.; Li, Z.; et al. Induction of Foxp3 and activation of Tregs by HSP gp96 for treatment of autoimmune diseases. iScience 2021, 24, 103445. [Google Scholar] [CrossRef]
- Liu, Y.; An, L.; Huang, R.; Xiong, J.; Yang, H.; Wang, X.; Zhang, X. Strategies to enhance CAR-T persistence. Biomark. Res. 2022, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Bartoló-Ibars, A.; Uribe-Herranz, M.; Muñoz-Sánchez, G.; Arnaldos-Pérez, C.; Ortiz-Maldonado, V.; Urbano-Ispizua, Á.; Pascal, M.; Juan, M. CAR-T after Stem Cell Transplantation in B-Cell Lymphoproliferative Disorders: Are They Really Autologous or Allogenic Cell Therapies? Cancers 2021, 13, 4664. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Oren, T.; Berke, G.; Kaufmann, Y. Cytotoxic T Cell Hybridomas: Generation and Characterization. Poxviruses 1982, 100, 11–18. [Google Scholar] [CrossRef]
- Harding, F.A.; Allison, J.P. CD28-B7 interactions allow the induction of CD8+ cytotoxic T lymphocytes in the absence of exogenous help. J. Exp. Med. 1993, 177, 1791–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [CrossRef]
- Tammana, S.; Huang, X.; Wong, M.; Milone, M.C.; Ma, L.; Levine, B.L.; June, C.H.; Wagner, J.E.; Blazar, B.R.; Zhou, X. 4-1BB and CD28 Signaling Plays a Synergistic Role in Redirecting Umbilical Cord Blood T Cells Against B-Cell Malignancies. Hum. Gene Ther. 2010, 21, 75–86. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofler, D.M.; Chmielewski, M.; Rappl, G.; Hombach, A.; Riet, T.; Schmidt, A.; Hombach, A.A.; Wendtner, C.M.; Abken, H. CD28 cost-imulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol. Ther. 2011, 19, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Bandukwala, H.S.; Wu, Y.; Feuerer, M.; Chen, Y.; Barboza, B.; Ghosh, S.; Stroud, J.C.; Benoist, C.; Mathis, D.; Rao, A.; et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 2011, 34, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Borde, M.; Heissmeyer, V.; Feuerer, M.; Lapan, A.D.; Stroud, J.C.; Bates, D.L.; Guo, L.; Han, A.; Ziegler, S.F.; et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006, 126, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Aringer, M.; Alarcón-Riquelme, M.E.; Clowse, M.; Pons-Estel, G.J.; Vital, E.M.; Dall’era, M. A glimpse into the future of systemic lupus erythematosus. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X221086719. [Google Scholar] [CrossRef]
- Radic, M.; Neeli, I.; Marion, T. Prospects for CAR T cell immunotherapy in autoimmune diseases: Clues from Lupus. Expert Opin. Biol. Ther. 2022, 22, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.-U.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell. Mol. Immunol. 2021, 18, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Boeltz, S.; Müller, F.; Kleyer, A.; Völkl, S.; Aigner, M.; Gary, R.; Kretschmann, S.; Simon, D.; Kharboutli, S.; et al. OP0279 CAR-T cell treatment of refractory systemic lupus erythematosus—Safety and preliminary efficacy data from the first four patients. Ann. Rheum. Dis. 2022, 81, 185. [Google Scholar] [CrossRef]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, J.; Cinquina, A.; Wang, Q.; Xu, H.; Zhang, Q.; Sun, L.; Chen, Q.; Xu, L.; Pinz, K.; et al. Treatment of Systemic Lupus Erythematosus using BCMA-CD19 Compound CAR. Stem Cell Rev. Rep. 2021, 17, 2120–2123. [Google Scholar] [CrossRef]
- Kim, Y.C.; Zhang, A.-H.; Su, Y.; Rieder, S.A.; Rossi, R.J.; Ettinger, R.A.; Pratt, K.P.; Shevach, E.M.; Scott, D.W. Engineered antigen-specific human regulatory T cells: Immunosuppression of FVIII-specific T- and B-cell responses. Blood 2015, 125, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Xun, L.; Zhang, R.; Hu, F.; Luan, J.; Lao, K.; Wang, X.; Gou, X. Stability and inhibitory function of Treg cells under inflammatory conditions in vitro. Exp. Ther. Med. 2020, 18, 2443–2450. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhang, R.; Shao, M.; Zhao, X.; Miao, M.; Chen, J.; Liu, J.; Zhang, X.; Zhang, X.; Jin, Y.; et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: A randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2019, 79, 141–149. [Google Scholar] [CrossRef]
- Zhan, Q.; Zhang, J.; Lin, Y.; Chen, W.; Fan, X.; Zhang, D. Pathogenesis and treatment of Sjogren’s syndrome: Review and update. Front. Immunol. 2023, 14, 1127417. [Google Scholar] [CrossRef]
- Nocturne, G. Actualités dans le syndrome de Sjögren primitif: Aspects cliniques et thérapeutiques [Sjögren’s syndrome update: Clinical and therapeutic aspects]. Rev. Med. Interne 2019, 40, 433–439. [Google Scholar] [CrossRef]
- Meng, H.; Sun, X.; Song, Y.; Zou, J.; An, G.; Jin, Z.; Yang, L. La/SSB chimeric autoantibody receptor modified NK92MI cells for targeted therapy of autoimmune disease. Clin. Immunol. 2018, 192, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Fayyaz, A.; Kurien, B.T.; Scofield, R.H. Autoantibodies in Sjögren’s Syndrome. Rheum. Dis. Clin. N. Am. 2016, 42, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; St Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Faculty Opinions recommendation of Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N. Engl. J. Med. 2011, 363, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Furuta, S.; Tervaert, J.W.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; et al. Rituximab versus cyclophosphamide in AN-CA-associated renal vasculitis: 2-year results of a randomised trial. Ann. Rheum. Dis. 2015, 74, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Garvey, B. Rituximab in the treatment of autoimmune haematological disorders. Br. J. Haematol. 2008, 141, 149–169. [Google Scholar] [CrossRef]
- Rodrigo, C.; Rajapakse, S.; Gooneratne, L. Rituximab in the treatment of autoimmune haemolytic anaemia. Br. J. Clin. Pharmacol. 2015, 79, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Du, Y. A comprehensive review of advanced drug delivery systems for the treatment of rheumatoid arthritis. Int. J. Pharm. 2023, 635, 122698. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Després, N.; Lapointe, E.; van der Heijden, A.; Lora, M.; Senshu, T.; van Venrooij, W.J.; Ménard, H.A. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Ther. 2004, 6, R142–R150. [Google Scholar] [CrossRef] [Green Version]
- Zanetakis, E.; Rigby, W.F.; Rubbert-Roth, A.; Peterfy, C.G.; Van Vollenhoven, R.F.; Stohl, W.; Hessey, E.; Chen, A.; Tyrrell, H.; Hackshaw, K.; et al. Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: The IMAGE trial. Ann. Rheum. Dis. 2011, 70, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wang, Y.; Yuan, Y.; Sun, J.; Liu, L.; Huang, D.; Hu, J.; Wang, M.; Li, S.; Song, W.; et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann. Rheum. Dis. 2021, 80, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Chen, Z. Cell-based therapies for rheumatoid arthritis: Opportunities and challenges. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X221100294. [Google Scholar] [CrossRef] [PubMed]
- Van Steendam, K.; Tilleman, K.; De Ceuleneer, M.; De Keyser, F.; Elewaut, D.; Deforce, D. Citrullinated vimentin as an important antigen in immune complexes from synovial fluid of rheumatoid arthritis patients with antibodies against citrullinated proteins. Arthritis Res. Ther. 2010, 12, R132. [Google Scholar] [CrossRef] [Green Version]
- Orvain, C.; Boulch, M.; Bousso, P.; Allanore, Y.; Avouac, J. Is There a Place for Chimeric Antigen Receptor-T Cells in the Treatment of Chronic Autoimmune Rheumatic Diseases? Arthritis Rheumatol. 2021, 73, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, A. Pathogenic roles of B lymphocytes in systemic sclerosis. Immunol. Lett. 2018, 195, 76–82. [Google Scholar] [CrossRef]
- Pillai, S. T and B lymphocytes in fibrosis and systemic sclerosis. Curr. Opin. Rheumatol. 2019, 31, 576–581. [Google Scholar] [CrossRef]
- Brown, M.; O’Reilly, S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin. Exp. Immunol. 2019, 195, 310–321. [Google Scholar] [CrossRef] [Green Version]
- Jordan, S.; Distler, J.H.; Maurer, B.; Huscher, D.; van Laar, J.M.; Allanore, Y.; Distler, O.; EUSTAR Rituximab study group. Effects and safety of rituximab in systemic sclerosis: An analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann. Rheum. Dis. 2015, 74, 1188–1194. [Google Scholar] [CrossRef]
- Daoussis, D.; Melissaropoulos, K.; Sakellaropoulos, G.; Antonopoulos, I.; Markatseli, T.E.; Simopoulou, T.; Georgiou, P.; Andonopoulos, A.P.; Drosos, A.A.; Sakkas, L.; et al. A multicenter, open-label, comparative study of B-cell depletion therapy with Rituximab for systemic sclerosis-associated interstitial lung disease. Semin. Arthritis Rheum. 2017, 46, 625–631. [Google Scholar] [CrossRef]
- Kuzumi, A.; Ebata, S.; Fukasawa, T.; Matsuda, K.M.; Kotani, H.; Yoshizaki-Ogawa, A.; Sato, S.; Yoshizaki, A. Long-term Outcomes After Rituximab Treatment for Patients With Systemic Sclerosis: Follow-up of the DESIRES Trial With a Focus on Serum Immunoglobulin Levels. JAMA Dermatol. 2023, 159, e226340. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Fernández, P.O.M.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Faculty Opinions recommendation of Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Faculty Opinions recommendation of Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2016, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Mitsdoerffer, M.; Di Liberto, G.; Dötsch, S.; Sie, C.; Wagner, I.; Pfaller, M.; Kreutzfeldt, M.; Fräßle, S.; Aly, L.; Knier, B.; et al. Formation and immunomodulatory function of meningeal B cell aggregates in progressive CNS autoimmunity. Brain 2021, 144, 1697–1710. [Google Scholar] [CrossRef]
- Weber, M.S.; Prod’homme, T.; Patarroyo, J.C.; Molnarfi, N.; Karnezis, T.; Lehmann-Horn, K.; Danilenko, D.M.; Eastham-Anderson, J.; Slavin, A.J.; Linington, C.; et al. B-cell activation influences T-cell polarization and outcome of an-ti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 2010, 68, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Häusler, D.; Häusser-Kinzel, S.; Feldmann, L.; Torke, S.; Lepennetier, G.; Bernard, C.C.A.; Zamvil, S.S.; Brück, W.; Lehmann-Horn, K.; Weber, M.S. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc. Natl. Acad. Sci. USA 2018, 115, 9773–9778. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Simic, M.; Sagan, S.A.; Shepherd, C.; Duecker, J.; Sobel, R.A.; Dandekar, R.; Wu, G.F.; Wu, W.; Pak, J.E.; et al. CAR-T Cell–Mediated B-Cell Depletion in Central Nervous System Autoimmunity. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200080. [Google Scholar] [CrossRef]
- Monson, N.L.; Cravens, P.D.; Frohman, E.M.; Hawker, K.; Racke, M.K. Effect of Rituximab on the Peripheral Blood and Cerebrospinal Fluid B Cells in Patients With Primary Progressive Multiple Sclerosis. Arch. Neurol. 2005, 62, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Mainero, C.; Louapre, C. Meningeal inflammation in multiple sclerosis: The key to the origin of cortical lesions? Neurology 2015, 85, 12–13. [Google Scholar] [CrossRef]
- Abramson, J.S.; McGree, B.; Noyes, S.; Plummer, S.; Wong, C.; Chen, Y.-B.; Palmer, E.; Albertson, T.; Ferry, J.A.; Arrillaga-Romany, I.C. Anti-CD19 CAR T Cells in CNS Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflamm. 2012, 9, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Zhang, A.H.; Yoon, J.; Culp, W.E.; Lees, J.R.; Wucherpfennig, K.W.; Scott, D.W. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J. Autoimmun. 2018, 92, 77–86. [Google Scholar] [CrossRef]
- De Paula Pohl, A.; Schmidt, A.; Zhang, A.H.; Maldonado, T.; Königs, C.; Scott, D.W. Engineered regulatory T cells expressing myelin-specific chimeric antigen receptors suppress EAE progression. Cell Immunol. 2020, 358, 104222. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.B.; McCune, J.M.; Miedema, F.; Moore, J.P.; Schuitemaker, H. HIV tropism and CD4+ T-cell depletion. Nat. Med. 2002, 8, 537. [Google Scholar] [CrossRef]
- Ghaleh, H.E.G.; Bolandian, M.; Dorostkar, R.; Jafari, A.; Pour, M.F. Concise review on optimized methods in production and transduction of lentiviral vectors in order to facilitate immunotherapy and gene therapy. Biomed. Pharmacother. 2020, 128, 110276. [Google Scholar] [CrossRef]
- Sedaghat, N.; Etemadifar, M. Inducing chimeric antigen receptor (CAR) regulatory T cells in-vivo: A novel concept for a potential feasible cure of demyelinating diseases. Mult. Scler. Relat. Disord. 2021, 57, 103341. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J.J.G.M. Myasthenia gravis. Nat. Rev. Dis. Prim. 2019, 5, 30. [Google Scholar] [CrossRef]
- Lazaridis, K.; Tzartos, S.J. Myasthenia Gravis: Autoantibody Specificities and Their Role in MG Management. Front. Neurol. 2020, 11, 596981. [Google Scholar] [CrossRef]
- Behin, A.; Le Panse, R. New Pathways and Therapeutic Targets in Autoimmune Myasthenia Gravis. J. Neuromuscul. Dis. 2018, 5, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Bagherieh, S.; Afshari-Safavi, A.; Vaheb, S.; Kiani, M.; Ghaffary, E.M.; Barzegar, M.; Shaygannejad, V.; Zabeti, A.; Mirmosayyeb, O. Worldwide prevalence of neuromyelitis optica spectrum disorder (NMOSD) and neuromyelitis optica (NMO): A systematic review and meta-analysis. Neurol. Sci. 2023, 44, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Wolbert, J.; Cheng, M.; Zu Horste, G.M.; Su, M.A. Deciphering immune mechanisms in chronic inflammatory demyelinating polyneuropathies. J. Clin. Investig. 2020, 5, e132411. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.J.; Wilkinson, J.; Coventry, B.J. Therapeutic options for chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review. BMC Neurol. 2014, 14, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuhlmüller, B.; Schneider, U.; González-González, J.B.; Feist, E. Disease specific autoantibodies in idiopathic inflammatory myopathies. Front. Neurol. 2019, 10, 438. [Google Scholar] [CrossRef] [Green Version]
- Xiong, A.; Yang, G.; Song, Z.; Xiong, C.; Liu, D.; Shuai, Y.; He, L.; Zhang, L.; Guo, Z.; Shuai, S. Rituximab in the treatment of immune-mediated necrotizing myopathy: A review of case reports and case series. Ther. Adv. Neurol. Disord. 2021, 14, 1756286421998918. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, Y.; Wang, M.; Leyang, X. The global incidence rate of pemphigus vulgaris: A systematic review and meta-analysis. Dermatology 2023, 239, 1–9. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Parvathaneni, K.; Scott, D.W. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2018, 2, 2332–2340. [Google Scholar] [CrossRef] [Green Version]
- Ohyama, B.; Nishifuji, K.; Chan, P.T.; Kawaguchi, A.; Yamashita, T.; Ishii, N.; Hamada, T.; Dainichi, T.; Koga, H.; Tsuruta, D.; et al. Epitope Spreading Is Rarely Found in Pemphigus Vulgaris by Large-Scale Longitudinal Study Using Desmoglein 2–Based Swapped Molecules. J. Investig. Dermatol. 2012, 132, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lundgren, D.K.; Mao, X.; Manfredo-Vieira, S.; Nunez-Cruz, S.; Williams, E.F.; Assenmacher, C.-A.; Radaelli, E.; Oh, S.; Wang, B.; et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J. Clin. Investig. 2020, 130, 6317–6324. [Google Scholar] [CrossRef]
- Schmidt, E.; Spindler, V.; Eming, R.; Amagai, M.; Antonicelli, F.; Baines, J.F.; Belheouane, M.; Bernard, P.; Borradori, L.; Caproni, M.; et al. Meeting Report of the Pathogenesis of Pemphigus and Pemphigoid Meeting in Munich, September 2016. J. Investig. Dermatol. 2017, 137, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, H.F.; Staser, K.W.; Nambudiri, V.E. Research Techniques Made Simple: CAR T-Cell Therapy. J. Investig. Dermatol. 2018, 138, 2501–2504.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colliou, N.; Picard, D.; Caillot, F.; Calbo, S.; Le Corre, S.; Lim, A.; Lemercier, B.; Le Mauff, B.; Maho-Vaillant, M.; Jacquot, S.; et al. Long-Term Remissions of Severe Pemphigus After Rituximab Therapy Are Associated with Prolonged Failure of Desmoglein B Cell Response. Sci. Transl. Med. 2013, 5, 175ra30. [Google Scholar] [CrossRef]
- Amagai, M. Pemphigus vulgaris and its active disease mouse model. Curr. Dir. Autoimmun. 2008, 10, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Nishifuji, K.; Amagai, M.; Kuwana, M.; Iwasaki, T.; Nishikawa, T. Detection of Antigen-Specific B Cells in Patients with Pemphigus Vulgaris by Enzyme-Linked Immunospot Assay: Requirement of T Cell Collaboration for Autoantibody Production. J. Investig. Dermatol. 2000, 114, 88–94. [Google Scholar] [CrossRef]
- Basu, S.; Volkov, J.; Nunez, D.; Fouch, M.; Stadanlick, J.; Binder, G.; Chang, D.; Hoffman, K.; Porter, D.; Abedi, M.; et al. Characterization of DSG3-CAART cells prior to & following adoptive transfer in mucosal Pemphigus Vulgaris. Hum. Gene Ther. 2022, 33, A123. [Google Scholar]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef] [Green Version]
- Tomaras, S.; Feist, E. Myositissyndrome [Myositis]. Inn. Med. 2023, 64, 152–163. [Google Scholar]
- Mavragani, C.P.; Spyridakis, E.G.; Koutsilieris, M. Adult-Onset Still’s Disease: From Pathophysiology to Targeted Therapies. Int. J. Inflamm. 2012, 2012, 879020. [Google Scholar] [CrossRef] [Green Version]
- Otte, M.L.; Tamang, R.L.; Papapanagiotou, J.; Ahmad, R.; Dhawan, P.; Singh, A.B. Mucosal healing and inflammatory bowel disease: Therapeutic implications and new targets. World J. Gastroenterol. 2023, 29, 1157–1172. [Google Scholar] [CrossRef]
- Gordon, H.; Rodger, B.; Lindsay, J.O.; Stagg, A.J. Recruitment and residence of intestinal T cells—Lessons for therapy in IBD. J. Crohn’s Colitis 2023, 17, jjad027. [Google Scholar] [CrossRef] [PubMed]
- Radziszewska, A.; Moulder, Z.; Jury, E.C.; Ciurtin, C. CD8+ T Cell Phenotype and Function in Childhood and Adult-Onset Connective Tissue Disease. Int. J. Mol. Sci. 2022, 23, 11431. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-Y.; Choi, B.; Sayeed, H.; Suh, C.-H.; Kim, Y.W.; Kim, H.-A.; Sohn, S. Characteristic patterns of HLA presentation and T cell differentiation in adult-onset Still’s disease. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418791284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Mu, W.; Zhao, Y.; Cheng, J.; Lin, H.; Ouyang, K.; Jia, X.; Liu, J.; Wei, Q.; Wang, M.; et al. T cells expressing CD5/CD7 bispecific chimeric antigen receptors with fully human heavy-chain-only domains mitigate tumor antigen escape. Signal Transduct. Target. Ther. 2022, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tan, Y.; Wang, G.; Deng, B.; Ling, Z.; Song, W.; Seery, S.; Zhang, Y.; Peng, S.; Xu, J.; et al. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. J. Clin. Oncol. 2021, 39, 3340–3351. [Google Scholar] [CrossRef]
- Breman, E.; Demoulin, B.; Agaugué, S.; Mauën, S.; Michaux, A.; Springuel, L.; Houssa, J.; Huberty, F.; Jacques-Hespel, C.; Marchand, C.; et al. Overcoming Target Driven Fratricide for T Cell Therapy. Front. Immunol. 2018, 9, 2940. [Google Scholar] [CrossRef] [PubMed]
- Moll, M.; Reinhold, U.; Kukel, S.; Abken, H.; Müller, R.; Oltermann, I.; Kreysel, H.W. CD7-negative helper T cells accumulate in inflammatory skin lesions. J. Investig. Dermatol. 1994, 102, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D.; Goronzy, J.J.; Weyand, C.M. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Investig. 1996, 97, 2027–2037. [Google Scholar] [CrossRef]
- Bishu, S.; Arsenescu, V.; Lee, E.Y.; Vargas, H.D.; De Villiers, W.J.; Arsenescu, R. Autoimmune enteropathy with a CD8+ CD7-T-cell small bowel intraepithelial lymphocytosis: Case report and literature review. BMC Gastroenterol. 2011, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Bo, X.-C.; Cao, H.-W.; Qian, C.; Wang, Z.; Li, B. Engineered T cells and their therapeutic applications in autoimmune diseases. Zool. Res. 2022, 43, 150–165. [Google Scholar] [CrossRef]
- Elinav, E.; Adam, N.; Waks, T.; Eshhar, Z. Amelioration of Colitis by Genetically Engineered Murine Regulatory T Cells Redirected by Antigen-Specific Chimeric Receptor. Gastroenterology 2009, 136, 1721–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elinav, E.; Waks, T.; Eshhar, Z. Redirection of Regulatory T Cells With Predetermined Specificity for the Treatment of Experimental Colitis in Mice. Gastroenterology 2008, 134, 2014–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, J.L.; Wright, G.P.; Stauss, H.J. Engineering Specificity and Function of Therapeutic Regulatory T Cells. Front. Immunol. 2017, 8, 1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freen-van Heeren, J.J. Using CRISPR to enhance T cell effector function for therapeutic applications. Cytokine X 2020, 3, 100049. [Google Scholar] [CrossRef]
- Blat, D.; Zigmond, E.; Alteber, Z.; Waks, T.; Eshhar, Z. Suppression of Murine Colitis and its Associated Cancer by Carcinoembryonic Antigen-Specific Regulatory T Cells. Mol. Ther. 2014, 22, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9, 59. [Google Scholar] [CrossRef]
- Syed, F.Z. Type 1 Diabetes Mellitus. Ann. Intern. Med. 2022, 175, ITC33–ITC48. [Google Scholar] [CrossRef]
- Fishman, S.; Lewis, M.D.; Siew, L.K.; De Leenheer, E.; Kakabadse, D.; Davies, J.; Ziv, D.; Margalit, A.; Karin, N.; Gross, G.; et al. Adoptive Transfer of mRNA-Transfected T Cells Redirected against Diabetogenic CD8 T Cells Can Prevent Diabetes. Mol. Ther. 2017, 25, 456–464. [Google Scholar] [CrossRef]
- Zhang, L.; Sosinowski, T.; Cox, A.R.; Cepeda, J.R.; Sekhar, N.S.; Hartig, S.M.; Miao, D.; Yu, L.; Pietropaolo, M.; Davidson, H.W. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II: Peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 2019, 96, 50–58. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Sobkowiak-Sobierajska, A.; Lindemans, C.; Sykora, T.; Wachowiak, J.; Dalle, J.H.; Bonig, H.; Gennery, A.; Lawitschka, A. Management of Chronic Graft-vs.-Host Disease in Children and Adolescents With ALL: Present Status and Model for a Personalised Management Plan. Front. Pediatr. 2022, 10, 808103. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Ando, M.; Kondo, T.; Ito, M.; Yoshimura, A. CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. J. Clin. Investig. 2020, 5, e136185. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Kang, C.H.; Choi, S.U.; Kim, Y.; Hwang, J.Y.; Jeong, H.G.; Park, C.H. A Chemical Switch System to Modulate Chimeric Antigen Receptor T Cell Activity through Proteolysis-Targeting Chimaera Technology. ACS Synth. Biol. 2020, 9, 987–992. [Google Scholar] [CrossRef]
- Fiorenza, S.; Ritchie, D.S.; Ramsey, S.D.; Turtle, C.J.; Roth, J.A. Value and affordability of CAR T-cell therapy in the United States. Bone Marrow Transplant. 2020, 55, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Wen, Q.; Zhong, J.F.; Zhang, C.; Zhang, X. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; He, J.; Liu, L.; Wang, J.; Wang, S.; Liu, L.; Ge, J.; Gao, L.; Gao, L.; Kong, P.; et al. Novel CD19 chimeric antigen receptor T cells manufactured next-day for acute lymphoblastic leukemia. Blood Cancer J. 2022, 12, 96. [Google Scholar] [CrossRef]
- Novartis. Charging towards the Next-Generation of CAR-T; Novartis: Basel, Switzerland, 2022. [Google Scholar]
- Bozza, M.; De Roia, A.; Correia, M.P.; Berger, A.; Tuch, A.; Schmidt, A.; Zörnig, I.; Jäger, D.; Schmidt, P.; Harbottle, R.P. A nonviral, nonintegrating DNA nanovector platform for the safe, rapid, and persistent manufacture of recombinant T cells. Sci. Adv. 2021, 7, eabf1333. [Google Scholar] [CrossRef]
- Sanber, K.; Savani, B.; Jain, T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: The diametric opposition of T cells. Br. J. Haematol. 2021, 195, 660–668. [Google Scholar] [CrossRef]
- Makita, S.; Yoshimura, K.; Tobinai, K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. 2017, 108, 1109–1118. [Google Scholar] [CrossRef] [Green Version]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Breslin, S. Cytokine-Release Syndrome: Overview and Nursing Implications. Clin. J. Oncol. Nurs. 2007, 11 (Suppl. S1), 37–42. [Google Scholar] [CrossRef] [Green Version]
- Stein-Merlob, A.F.; Rothberg, M.V.; Holman, P.; Yang, E.H. Immunotherapy-Associated Cardiotoxicity of Immune Checkpoint Inhibitors and Chimeric Antigen Receptor T Cell Therapy: Diagnostic and Management Challenges and Strategies. Curr. Cardiol. Rep. 2021, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Sdrimas, K.; Diaz-Paez, M.; Camargo, J.F.; Lekakis, L.J. Progressive multifocal leukoencephalopathy after CAR T therapy. Int. J. Hematol. 2020, 112, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, H.; Rurik, J.G.; Epstein, J.A. CAR-based therapies: Opportunities for immuno-medicine beyond cancer. Nat. Metab. 2022, 4, 163–169. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Report or Trial Registry Number | Conditioning Treatment | CAR-Construct | Dosage of CAR Cells (Cells/kg Body Weight) | Overall Outcomes (or Primary Outcome Measures) | References |
|---|---|---|---|---|---|---|
| SLE (+ LN) | report | fludarabine | anti-CD19 CAR T | 1.1 × 106 | - SELENA score decreased from 16 to 0 - dsDNA, C3, C4 levels normalized - patients achieved LLDAS - glucocorticoids were discontinued | [63,64] |
| SLE | report | fludarabine + cyclophosphamide | anti-CD19 CAR T | 1 × 106 | - SLE remission according to DORIS criteria occurred within 3 months - SLEDAI median range became 0 - drug-free remission up to 8 months | [65] |
| SLE (+ stage IV DLBCL) | report | fludarabine + cyclophosphamide | anti-BCMA/CD19 compound CAR T (CD137 co-stimulation) | 5.3 × 106 | - disease remission after 20 weeks - ANA was negative for 37 weeks | [66] |
| SLE | NCT03030976 | cyclophosphamide | anti-CD19 CAR T (4-1BB co-stimulation) | 106–107 | - safety - efficacy | |
| SLE | NCT05030779 | not available | anti-BCMA/CD19 compound CAR T | 1–4 × 106 | - safety - efficacy | |
| SLE | NCT05474885 | not available | anti-BCMA/CD19 compound CAR T | not available | - adverse effects | |
| SLE | NCT05765006 | not available | anti-CD19 CAR T (Relma-cel) | 15–150 × 106 | - safety - efficacy - pharmacokinetics - pharmacodynamics | |
| SS | NCT05085431 | not available | anti-BCMA/CD19 compound CAR T | 1–4 × 106 | - safety - efficacy - dose-limiting toxicity - adverse effects | |
| ANCA-associated vasculitis, AIHA (+ POEMS syndrome and amyloidosis) | NCT05263817 | not available | anti-BCMA/CD19 compound CAR T | not available | - dose-limiting toxicity - safety - tolerability | |
| SSc | NCT05085444 | not available | anti-BCMA/CD19 compound CAR T | not available | - dose-limiting toxicity - safety - tolerability | |
| MG | NCT04146051 | not available | anti-BCMA CAR T (Descartes-08) | not available | - safety - tolerability - preliminary efficacy of a repeated dosing schedule of Descartes-08 CAR T | |
| MG, NMOSD, CIDP, IMNM | NCT04561557 | fludarabine + cyclophosphamide | anti-BCMA CAR T (CT103A) | 0.25–1 × 106 | - safety - dose-limiting toxicity - efficacy | |
| MG | NCT05451212 | cyclophosphamide (+/− fludarabine) | anti-MuSK CAAR T | not available | - adverse events - dose-limiting toxicity | |
| PV | report | anti-DSG3 CAAR T | up to 2.5 × 109 | - no dose-limiting adverse effects - transient improvements in clinical disease activity and antibody levels with a duration of two months | [127] | |
| PV | NCT04422912 | intravenous immunoglobulin, cyclophosphamide (+/−fludarabine) | anti-DSG3 CAAR T | not available | - adverse events - dose-limiting toxicity | |
| CD, UC, DM, AOSD | NCT05239702 | not available | anti-CD7 CAR T | not available | - dose-limiting toxicity - safety - tolerability | |
| IBD, GvHD (+ functional gastrointestinal disorders) | NCT03369353 (PREDICT trial) | not available | non-defined CAR T | not available | - applying a systems-biology approach to enable precision diagnostics for the key immunologic outcomes for the investigated disorders - investigation of the immunology of auto- and alloimmune gastrointestinal disorders - revealing the immune manifestations after CAR T therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Műzes, G.; Sipos, F. CAR-Based Therapy for Autoimmune Diseases: A Novel Powerful Option. Cells 2023, 12, 1534. https://doi.org/10.3390/cells12111534
Műzes G, Sipos F. CAR-Based Therapy for Autoimmune Diseases: A Novel Powerful Option. Cells. 2023; 12(11):1534. https://doi.org/10.3390/cells12111534
Chicago/Turabian StyleMűzes, Györgyi, and Ferenc Sipos. 2023. "CAR-Based Therapy for Autoimmune Diseases: A Novel Powerful Option" Cells 12, no. 11: 1534. https://doi.org/10.3390/cells12111534
APA StyleMűzes, G., & Sipos, F. (2023). CAR-Based Therapy for Autoimmune Diseases: A Novel Powerful Option. Cells, 12(11), 1534. https://doi.org/10.3390/cells12111534