

TMEM106B Puncta Is Increased in Multiple Sclerosis Plaques, and Reduced Protein in Mice Results in Delayed Lipid Clearance Following CNS Injury


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Materials and Methods
2.1. Isolation of Insoluble Proteins from Human White Matter
2.2. Protein Extraction and Sample Preparation
2.3. Sample Desalting
2.4. LC-MS/MS Acquisition
2.5. Proteomics Data Analysis
2.6. Stains and Antibodies
2.6.1. Myelin Stain
2.6.2. Antibodies
2.7. Immunostaining and Analysis of Human Brain Sections
2.8. Mice
2.9. MOG-Induced EAE
2.10. Cuprizone Treatment
2.11. Oil Red O Staining
2.12. Histologic Grading
2.13. Statistical Analysis
2.14. Data Availability
3. Results
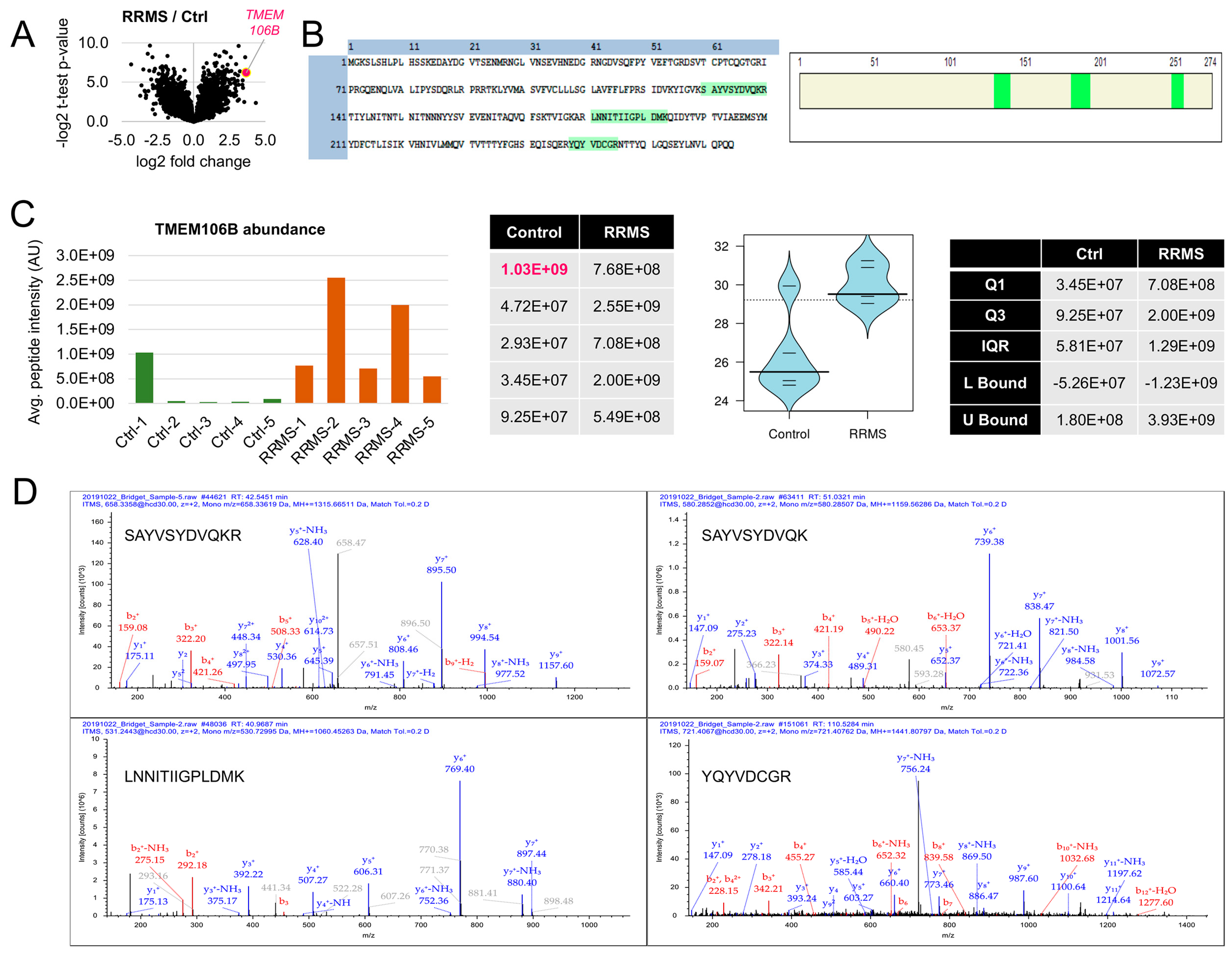
3.1. TMEM106B Is Elevated in Insoluble Protein Pellets Isolated from the White Matter Plaques of Individuals with RRMS
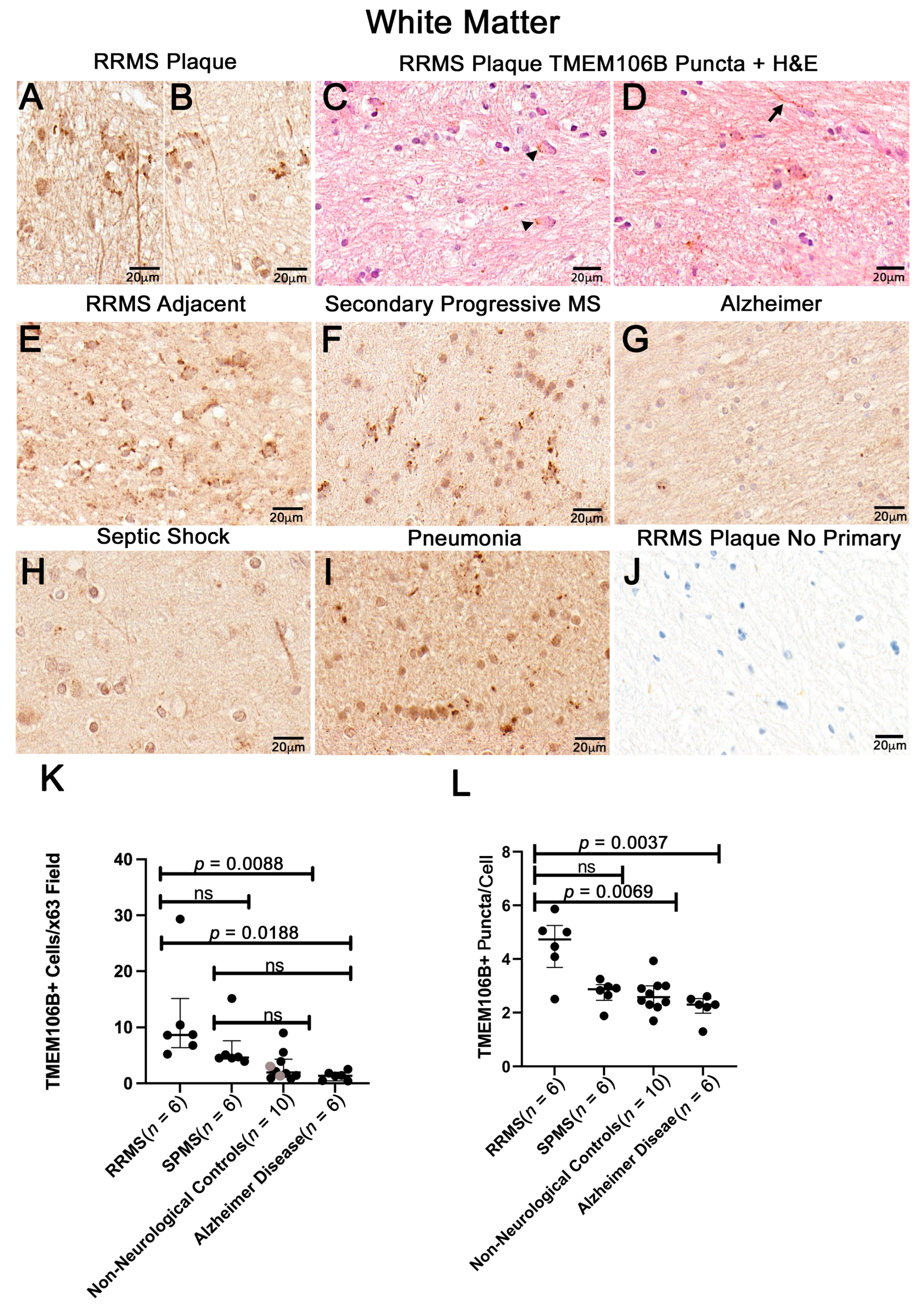
3.2. White Matter Plaques of Individuals with RRMS Show Increased TMEM106B+ Puncta Relative to the White Matter of Controls

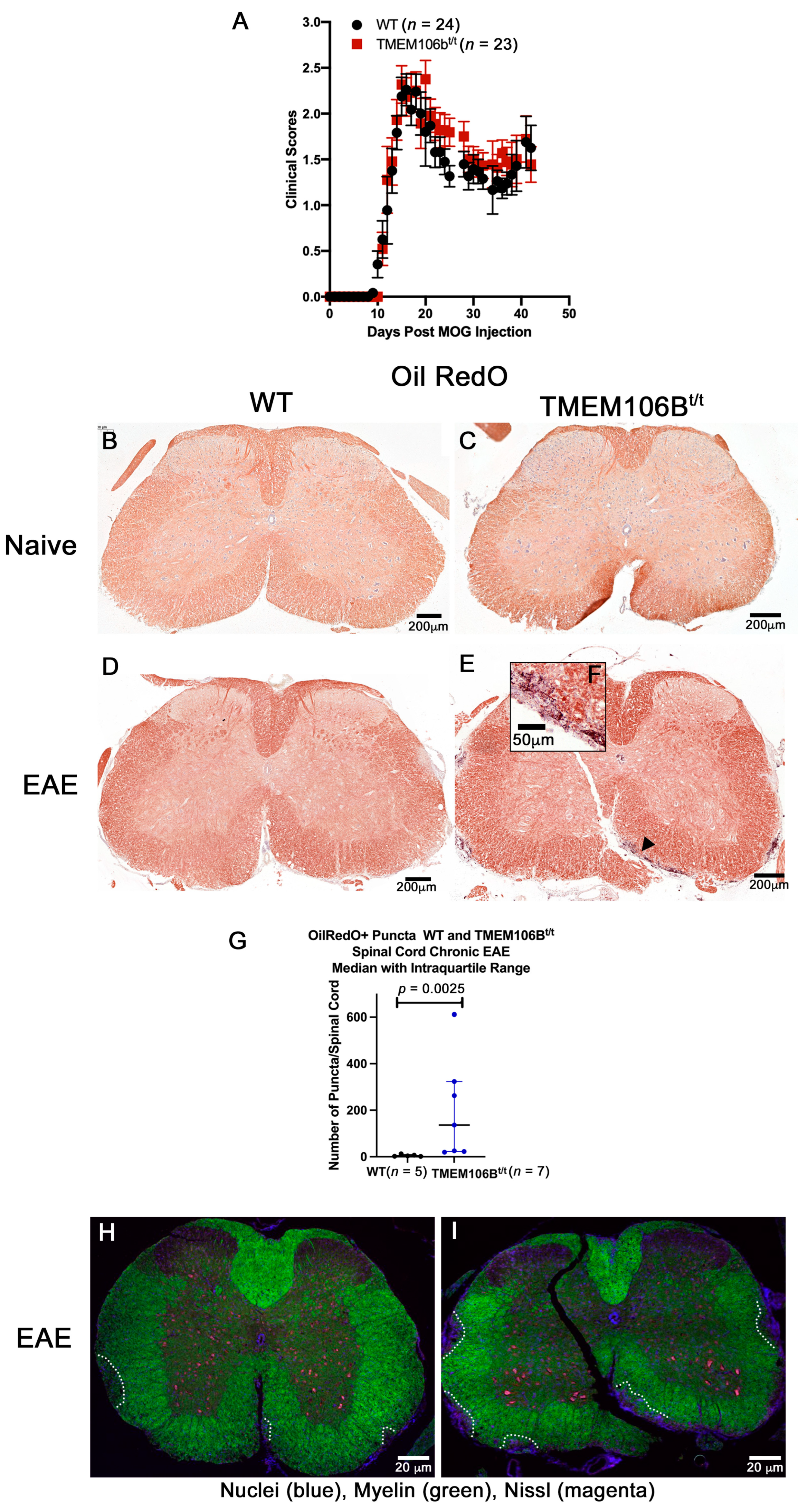
3.3. Naïve WT and TMEM106Bt/t Mice Exhibit Similar Myelination and CNS Architecture at the Corpus Callosum and Spinal Cord
3.4. TMEM106Bt/t Mice Have More OilRedO+ Puncta, Axonal Damage, and Demyelination during Chronic EAE

3.5. WT and TMEM106Bt/t Mice Have Equivalent OilRedO+ Staining during Late Acute EAE



3.6. TMEM106Bt/t Mice Have OilRedO+ Deposits in the Corpus Callosum at 2 and 3 Weeks Post-Cuprizone Withdrawal

4. Discussion

Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef]
- Popescu, B.F.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Continuum 2013, 19, 901–921. [Google Scholar] [CrossRef]
- Sanders, V.; Conrad, A.J.; Tourtellotte, W.W. On classification of post-mortem multiple sclerosis plaques for neuroscientists. J. Neuroimmunol. 1993, 46, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Raine, C.S. Multiple Scelrosis: Clinical and pathogenetic basis. BMJ 1997, 314, 1228. [Google Scholar]
- Boyle, E.A.; McGeer, P.L. Cellular immune response in multiple sclerosis plaques. Am. J. Pathol. 1990, 137, 575–584. [Google Scholar]
- Lassmann, H.; Raine, C.S.; Antel, J.; Prineas, J.W. Immunopathology of multiple sclerosis: Report on an international meeting held at the Institute of Neurology of the University of Vienna. J. Neuroimmunol. 1998, 86, 213–217. [Google Scholar] [CrossRef]
- Junker, A.; Wozniak, J.; Voigt, D.; Scheidt, U.; Antel, J.; Wegner, C.; Bruck, W.; Stadelmann, C. Extensive subpial cortical demyelination is specific to multiple sclerosis. Brain Pathol. 2020, 30, 641–652. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Bruck, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammel, G.; Zivkovic, S.; Ayazi, M.; Ren, Y. Consequences and mechanisms of myelin debris uptake and processing by cells in the central nervous system. Cell. Immunol. 2022, 380, 104591. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, O.B.; Adams, C.W. Cellular lipid inclusions in the white matter in multiple sclerosis. Nature 1971, 233, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Luningschror, P.; Werner, G.; Stroobants, S.; Kakuta, S.; Dombert, B.; Sinske, D.; Wanner, R.; Lullmann-Rauch, R.; Wefers, B.; Wurst, W.; et al. The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep. 2020, 30, 3506–3519.e6. [Google Scholar] [CrossRef] [Green Version]
- Feng, T.; Sheng, R.R.; Sole-Domenech, S.; Ullah, M.; Zhou, X.; Mendoza, C.S.; Enriquez, L.C.M.; Katz, I.I.; Paushter, D.H.; Sullivan, P.M.; et al. A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 2020, 143, 2255–2271. [Google Scholar] [CrossRef]
- Nicholson, A.M.; Finch, N.A.; Wojtas, A.; Baker, M.C.; Perkerson, R.B., 3rd; Castanedes-Casey, M.; Rousseau, L.; Benussi, L.; Binetti, G.; Ghidoni, R.; et al. TMEM106B p.T185S regulates TMEM106B protein levels: Implications for frontotemporal dementia. J. Neurochem. 2013, 126, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Chen-Plotkin, A.S.; Unger, T.L.; Gallagher, M.D.; Bill, E.; Kwong, L.K.; Volpicelli-Daley, L.; Busch, J.I.; Akle, S.; Grossman, M.; Van Deerlin, V.; et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J. Neurosci. 2012, 32, 11213–11227. [Google Scholar] [CrossRef] [Green Version]
- Busch, J.I.; Martinez-Lage, M.; Ashbridge, E.; Grossman, M.; Van Deerlin, V.M.; Hu, F.; Lee, V.M.; Trojanowski, J.Q.; Chen-Plotkin, A.S. Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol. Commun. 2013, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [Green Version]
- Stagi, M.; Klein, Z.A.; Gould, T.J.; Bewersdorf, J.; Strittmatter, S.M. Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol. Cell. Neurosci. 2014, 61, 226–240. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, B.M.; Lang, C.M.; Hogl, S.; Tahirovic, S.; Orozco, D.; Rentzsch, K.; Lichtenthaler, S.F.; Hoogenraad, C.C.; Capell, A.; Haass, C.; et al. The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 2014, 33, 450–467. [Google Scholar] [CrossRef] [PubMed]
- Perez-Canamas, A.; Takahashi, H.; Lindborg, J.A.; Strittmatter, S.M. Fronto-temporal dementia risk gene TMEM106B has opposing effects in different lysosomal storage disorders. Brain Commun. 2021, 3, fcaa200. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Lacrampe, A.; Hu, F. Physiological and pathological functions of TMEM106B: A gene associated with brain aging and multiple brain disorders. Acta Neuropathol. 2021, 141, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Nicholson, A.M.; Ren, Y.; Brooks, M.; Jiang, P.; Zuberi, A.; Phuoc, H.N.; Perkerson, R.B.; Matchett, B.; Parsons, T.M.; et al. Loss of TMEM106B leads to myelination deficits: Implications for frontotemporal dementia treatment strategies. Brain 2020, 143, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Nicholson, A.M.; Ren, Y.; Koga, S.; Nguyen, H.P.; Brooks, M.; Qiao, W.; Quicksall, Z.S.; Matchett, B.; Perkerson, R.B.; et al. Loss of Tmem106b leads to cerebellum Purkinje cell death and motor deficits. Brain Pathol. 2021, 31, e12945. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, G.K.; Morell, P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001, 11, 107–116. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S.S. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005, 26, 565–571. [Google Scholar] [CrossRef]
- Farooqi, N.; Gran, B.; Constantinescu, C.S. Are current disease-modifying therapeutics in multiple sclerosis justified on the basis of studies in experimental autoimmune encephalomyelitis? J. Neurochem. 2010, 115, 829–844. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, S.G.; Davies, P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 5827–5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, Z.A.; Takahashi, H.; Ma, M.; Stagi, M.; Zhou, M.; Lam, T.T.; Strittmatter, S.M. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron 2017, 95, 281–296.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, R.C.; Ray, A.K.; Johndrow, C.T.; Guzik, H.; Burek, D.; de Frutos, P.G.; Shafit-Zagardo, B. Targeted GAS6 delivery to the CNS protects axons from damage during experimental autoimmune encephalomyelitis. J. Neurosci. 2014, 34, 16320–16335. [Google Scholar] [CrossRef] [Green Version]
- Sidman, R.L.; Angevine, A.J.; Pierce, E.T. Atlas of Mouse Brain and Spinal Cord; Harvard University Press: Cambridge, MA, USA, 1971. [Google Scholar]
- DuBois, J.C.; Ray, A.K.; Gruber, R.C.; Zhang, Y.; Aflakpui, R.; Macian-Juan, F.; Shafit-Zagardo, B. Akt3-Mediated Protection Against Inflammatory Demyelinating Disease. Front. Immunol. 2019, 10, 1738. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Illouz, T.; Madar, R.; Biragyn, A.; Okun, E. Restoring microglial and astroglial homeostasis using DNA immunization in a Down Syndrome mouse model. Brain Behav. Immun. 2019, 75, 163–180. [Google Scholar] [CrossRef]
- Fiore, N.T.; Yin, Z.; Guneykaya, D.; Gauthier, C.D.; Hayes, J.P.; D’Hary, A.; Butovsky, O.; Moalem-Taylor, G. Sex-specific transcriptome of spinal microglia in neuropathic pain due to peripheral nerve injury. Glia 2022, 70, 675–696. [Google Scholar] [CrossRef]
- Hayashi, C.; Suzuki, N.; Takahashi, R.; Akazawa, C. Development of type I/II oligodendrocytes regulated by teneurin-4 in the murine spinal cord. Sci. Rep. 2020, 10, 8611. [Google Scholar] [CrossRef]
- Nicholson, A.M.; Rademakers, R. What we know about TMEM106B in neurodegeneration. Acta Neuropathol. 2016, 132, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boven, L.A.; Van Meurs, M.; Van Zwam, M.; Wierenga-Wolf, A.; Hintzen, R.Q.; Boot, R.G.; Aerts, J.M.; Amor, S.; Nieuwenhuis, E.E.; Laman, J.D. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain 2006, 129, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, F.; Lehmann-Horn, K.; Shen, Y.A.; Rankin, K.A.; Stebbins, K.J.; Lorrain, D.S.; Pekarek, K.; Sagan, A.S.; Xiao, L.; Teuscher, C.; et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 2016, 5, e18246. [Google Scholar] [CrossRef] [PubMed]
- Wujek, J.R.; Bjartmar, C.; Richer, E.; Ransohoff, R.M.; Yu, M.; Tuohy, V.K.; Trapp, B.D. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J. Neuropathol. Exp. Neurol. 2002, 61, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Dai, X.; Chen, J.; Zhao, J.; Xu, M.; Zhang, L.; Yang, B.; Zhang, W.; Rocha, M.; Nakao, T.; et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Chen, C.; Li, Q.; Darrow, A.L.; Wang, Y.; Derian, C.K.; Yang, J.; de Garavilla, L.; Andrade-Gordon, P.; Damiano, B.P. Mer receptor tyrosine kinase signaling participates in platelet function. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1118–1123. [Google Scholar] [CrossRef]
- Fenn, A.M.; Hall, J.C.; Gensel, J.C.; Popovich, P.G.; Godbout, J.P. IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: Consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J. Neurosci. 2014, 34, 8904–8917. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Quirie, A.; Demougeot, C.; Bertrand, N.; Mossiat, C.; Garnier, P.; Marie, C.; Prigent-Tessier, A. Effect of stroke on arginase expression and localization in the rat brain. Eur. J. Neurosci. 2013, 37, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Truettner, J.S.; Bramlett, H.M.; Dietrich, W.D. Posttraumatic therapeutic hypothermia alters microglial and macrophage polarization toward a beneficial phenotype. J. Cereb. Blood Flow Metab. 2017, 37, 2952–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, O.A.; Zheng, Y.; Murphy, K.; Huang, M.; Hu, F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum. Mol. Genet. 2013, 22, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, C.M.; Fellerer, K.; Schwenk, B.M.; Kuhn, P.H.; Kremmer, E.; Edbauer, D.; Capell, A.; Haass, C. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J. Biol. Chem. 2012, 287, 19355–19365. [Google Scholar] [CrossRef] [Green Version]
- Arrant, A.E.; Nicholson, A.M.; Zhou, X.; Rademakers, R.; Roberson, E.D. Partial Tmem106b reduction does not correct abnormalities due to progranulin haploinsufficiency. Mol. Neurodegener. 2018, 13, 32. [Google Scholar] [CrossRef]
- Feng, T.; Luan, L.; Katz, I.I.; Ullah, M.; Van Deerlin, V.M.; Trojanowski, J.Q.; Lee, E.B.; Hu, F. TMEM106B deficiency impairs cerebellar myelination and synaptic integrity with Purkinje cell loss. Acta Neuropathol. Commun. 2022, 10, 33. [Google Scholar] [CrossRef]
- Ruther, B.J.; Scheld, M.; Dreymueller, D.; Clarner, T.; Kress, E.; Brandenburg, L.O.; Swartenbroekx, T.; Hoornaert, C.; Ponsaerts, P.; Fallier-Becker, P.; et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia 2017, 65, 1900–1913. [Google Scholar] [CrossRef]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Levine, T.P. TMEM106B in humans and Vac7 and Tag1 in yeast are predicted to be lipid transfer proteins. Proteins 2021, 90, 164–175. [Google Scholar] [CrossRef]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif-crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Haidar, M.; Loix, M.; Vanherle, S.; Dierckx, T.; Vangansewinkel, T.; Gervois, P.; Wolfs, E.; Lambrichts, I.; Bogie, J.F.J.; Hendriks, J.J.A. Targeting lipophagy in macrophages improves repair in multiple sclerosis. Autophagy 2022, 18, 2697–2710. [Google Scholar] [CrossRef]
- Shimabukuro, M.K.; Langhi, L.G.; Cordeiro, I.; Brito, J.M.; Batista, C.M.; Mattson, M.P.; Mello Coelho, V. Lipid-laden cells differentially distributed in the aging brain are functionally active and correspond to distinct phenotypes. Sci. Rep. 2016, 6, 23795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Lim, L.; Song, J. TMEM106B, a risk factor for FTLD and aging, has an intrinsically disordered cytoplasmic domain. PLoS ONE 2018, 13, e0205856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.; Xiang, X.; Wang, J.; Lee, C.; Arakhamia, T.; Simjanoska, M.; Wang, C.; Carlomagno, Y.; Zhang, G.; Dhingra, S.; et al. Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 2022, 185, 1346–1355.e15. [Google Scholar] [CrossRef]
- Jiang, Y.X.; Cao, Q.; Sawaya, M.R.; Abskharon, R.; Ge, P.; DeTure, M.; Dickson, D.W.; Fu, J.Y.; Ogorzalek Loo, R.R.; Loo, J.A.; et al. Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 2022, 605, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Arseni, D.; Bacioglu, M.; Huang, M.; Lovestam, S.; Shi, Y.; Yang, Y.; Zhang, W.; Kotecha, A.; Garringer, H.J.; et al. Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 2022, 605, 310–314. [Google Scholar] [CrossRef]
- Cykowski, M.D.; Arumanayagam, A.S.; Powell, S.Z.; Rivera, A.L.; Abner, E.L.; Roman, G.C.; Masdeu, J.C.; Nelson, P.T. Patterns of amygdala region pathology in LATE-NC: Subtypes that differ with regard to TDP-43 histopathology, genetic risk factors, and comorbid pathologies. Acta Neuropathol. 2022, 143, 531–545. [Google Scholar] [CrossRef]
- Fan, Y.; Zhao, Q.; Xia, W.; Tao, Y.; Yu, W.; Chen, M.; Liu, Y.; Zhao, J.; Shen, Y.; Sun, Y.; et al. Generic amyloid fibrillation of TMEM106B in patient with Parkinson’s disease dementia and normal elders. Cell Res. 2022, 32, 585–588. [Google Scholar] [CrossRef]
- Simons, C.; Dyment, D.; Bent, S.J.; Crawford, J.; D’Hooghe, M.; Kohlschutter, A.; Venkateswaran, S.; Helman, G.; Poll-The, B.T.; Makowski, C.C.; et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain 2017, 140, 3105–3111. [Google Scholar] [CrossRef] [Green Version]
- Van Deerlin, V.M.; Sleiman, P.M.; Martinez-Lage, M.; Chen-Plotkin, A.; Wang, L.S.; Graff-Radford, N.R.; Dickson, D.W.; Rademakers, R.; Boeve, B.F.; Grossman, M.; et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 2010, 42, 234–239. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tropea, T.F.; Mak, J.; Guo, M.H.; Xie, S.X.; Suh, E.; Rick, J.; Siderowf, A.; Weintraub, D.; Grossman, M.; Irwin, D.; et al. TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann. Neurol. 2019, 85, 801–811. [Google Scholar] [CrossRef] [PubMed]
- van der Zee, J.; Van Langenhove, T.; Kleinberger, G.; Sleegers, K.; Engelborghs, S.; Vandenberghe, R.; Santens, P.; Van den Broeck, M.; Joris, G.; Brys, J.; et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 2011, 134, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafit-Zagardo, B.; Sidoli, S.; Goldman, J.E.; DuBois, J.C.; Corboy, J.R.; Strittmatter, S.M.; Guzik, H.; Edema, U.; Arackal, A.G.; Botbol, Y.M.; et al. TMEM106B Puncta Is Increased in Multiple Sclerosis Plaques, and Reduced Protein in Mice Results in Delayed Lipid Clearance Following CNS Injury. Cells 2023, 12, 1734. https://doi.org/10.3390/cells12131734
Shafit-Zagardo B, Sidoli S, Goldman JE, DuBois JC, Corboy JR, Strittmatter SM, Guzik H, Edema U, Arackal AG, Botbol YM, et al. TMEM106B Puncta Is Increased in Multiple Sclerosis Plaques, and Reduced Protein in Mice Results in Delayed Lipid Clearance Following CNS Injury. Cells. 2023; 12(13):1734. https://doi.org/10.3390/cells12131734
Chicago/Turabian StyleShafit-Zagardo, Bridget, Simone Sidoli, James E. Goldman, Juwen C. DuBois, John R. Corboy, Stephen M. Strittmatter, Hillary Guzik, Ukuemi Edema, Anita G. Arackal, Yair M. Botbol, and et al. 2023. "TMEM106B Puncta Is Increased in Multiple Sclerosis Plaques, and Reduced Protein in Mice Results in Delayed Lipid Clearance Following CNS Injury" Cells 12, no. 13: 1734. https://doi.org/10.3390/cells12131734
APA StyleShafit-Zagardo, B., Sidoli, S., Goldman, J. E., DuBois, J. C., Corboy, J. R., Strittmatter, S. M., Guzik, H., Edema, U., Arackal, A. G., Botbol, Y. M., Merheb, E., Nagra, R. M., & Graff, S. (2023). TMEM106B Puncta Is Increased in Multiple Sclerosis Plaques, and Reduced Protein in Mice Results in Delayed Lipid Clearance Following CNS Injury. Cells, 12(13), 1734. https://doi.org/10.3390/cells12131734