
Global Analysis of Transcription Start Sites and Enhancers in Endometrial Stromal Cells and Differences Associated with Endometriosis

 ,
,
Abstract
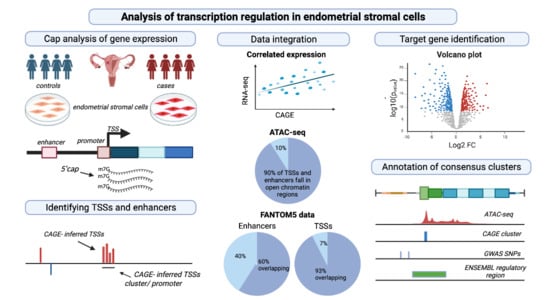
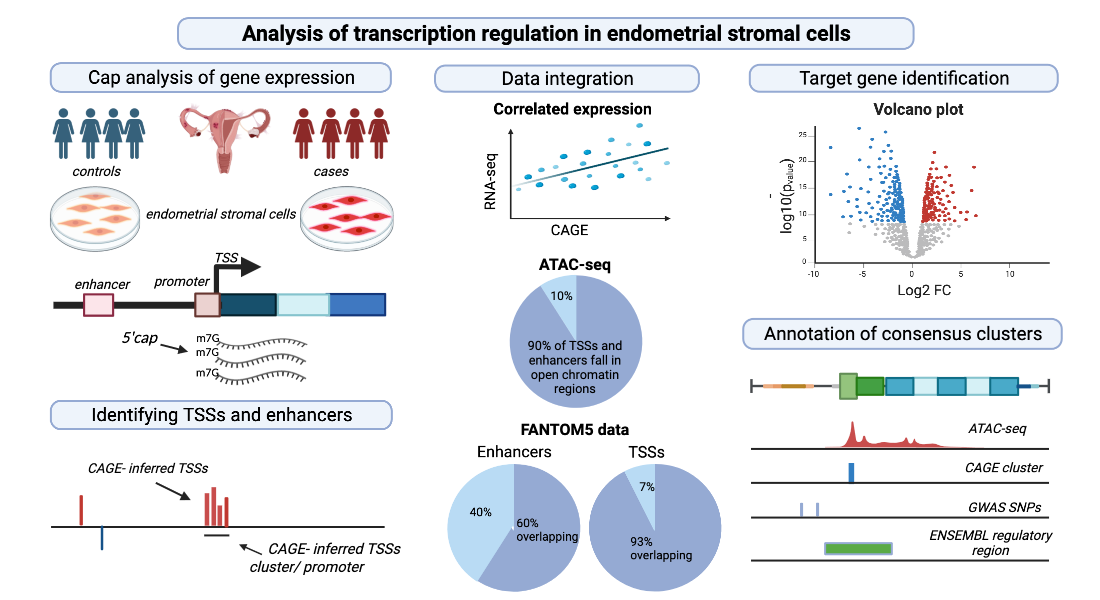
:
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. CAGE Sequencing
2.3. Identification of CAGE-Defined Transcription Start Sites (CTSS) and Tag Clusters (TC)
2.4. Correlation of Tag Counts within and between Protocols
2.5. Merging Data from In-House and Commercial Protocols
2.6. Promoter Shapes
2.7. Enhancer Identification
2.8. Interaction of Transcription Start Sites and Enhancers
2.9. Differential Promoter and Enhancer Expression
2.10. RNA Extraction and Sequencing
2.11. Correlation of CAGE-Seq Data with RNA-Seq Data
2.12. ATAC-Seq Using the Omni-ATAC Protocol from Actively Growing Endometrial Stromal Cells
2.13. Overlap of ESC CAGE Tag Clusters with ESC ATAC-Seq Peaks
2.14. Overlap of ESC Consensus Clusters with FANTOM5 and ENSEMBL Databases
2.15. Overlap of ESC Consensus Clusters with Endometriosis GWAS Signals
2.16. Pathway Analysis
3. Results
3.1. Comparing Quality of the Commercial and In-House CAGE Sequencing Protocols
3.2. Genomic Distribution of CAGE-Defined Transcription Start Sites (CTSS)
3.3. Identification of ESC Promoter Elements
3.4. Concordance between Sequencing Technologies
3.5. Transcriptional and Regulatory Elements Are Supported by ATAC-Seq Data
3.6. Differential Expression of Promoters between Endometriosis Cases and Controls
3.7. Predicted Enhancers and Differential Expression Analysis between Endometriosis Cases and Controls
3.8. ESC-Specific Promoters and Enhancers
3.9. Comparison of ESC CAGE Data with ENSEMBL TSS Annotation
3.10. Evidence of Transcriptional Elements in Endometriosis Risk Regions
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, J.; Salamonsen, L.A.; Winship, A.; Menkhorst, E.; Nie, G.; Gargett, C.E.; Dimitriadis, E. Fertile ground: Human endometrial programming and lessons in health and disease. Nat. Rev. Endocrinol. 2016, 12, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E.; Hayashi, K.; Hu, J.; Carpenter, K.D. Comparative Developmental Biology of the Mammalian Uterus. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2005; Volume 68, pp. 85–122. [Google Scholar] [CrossRef]
- Fung, J.N.; Mortlock, S.; Girling, J.E.; Holdsworth-Carson, S.J.; Teh, W.T.; Zhu, Z.; Lukowski, S.W.; McKinnon, B.D.; McRae, A.; Yang, J.; et al. Genetic regulation of disease risk and endometrial gene expression highlights potential target genes for endometriosis and polycystic ovarian syndrome. Sci. Rep. 2018, 8, 11424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortlock, S.; Kendarsari, I.R.; Fung, J.N.; Gibson, G.; Yang, F.; Restuadi, R.; Girling, E.J.; Holdsworth-Carson, S.J.; Teh, W.T.; Lukowski, S.W.; et al. Tissue specific regulation of transcription in endometrium and association with disease. Hum. Reprod. 2020, 35, 377–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Nilufer, R.; Banasik, K.; Paraskevi, C.; Danning, R.; Genevieve, G.; Ayush, G.; MacGregor, S.; Mortlock, S.; Sapkota, Y.; Schork, J.A.; et al. Large-scale genome-wide association meta-analysis of endometriosis reveals 13 novel loci and genetically-associated comorbidity with other pain conditions. bioRxiv 2018, 406967. [Google Scholar]
- Tanos, P.; Dimitriou, S.; Gullo, G.; Tanos, V. Biomolecular and Genetic Prognostic Factors That Can Facilitate Fertility-Sparing Treatment (FST) Decision Making in Early Stage Endometrial Cancer (ES-EC): A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2653. [Google Scholar] [CrossRef]
- Dyson, M.; Roqueiro, D.; Monsivais, D.; Ercan, C.M.; Pavone, M.E.; Brooks, D.C.; Kakinuma, T.; Ono, M.; Jafari, N.; Dai, Y.; et al. Genome-Wide DNA Methylation Analysis Predicts an Epigenetic Switch for GATA Factor Expression in Endometriosis. PLoS Genet. 2014, 10, e1004158. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef] [Green Version]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M.; et al. CAGE: Cap analysis of gene expression. Nat. Methods 2006, 3, 211–222. [Google Scholar] [CrossRef]
- Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Baillie, K.J.; de Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar]
- American Society for Reproductive Medicine. Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil. Steril. 1997, 67, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Burla, L.; Scheiner, D.; Samartzis, E.P.; Seidel, S.; Eberhard, M.; Fink, D.; Boss, A.; Imesch, P. The ENZIAN score as a preoperative MRI-based classification instrument for deep infiltrating endometriosis. Arch. Gynecol. Obstet. 2019, 300, 109–116. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, B.D.; Evers, J.; Bersinger, N.A.; Mueller, M. Induction of the Neurokinin 1 Receptor by TNFα in Endometriotic Tissue Provides the Potential for Neurogenic Control Over Endometriotic Lesion Growth. J. Clin. Endocrinol. Metab. 2013, 98, 2469–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, B.D.; Lukowski, S.W.; Mortlock, S.; Crawford, J.; Atluri, S.; Subramaniam, S.; Johnston, R.L.; Nirgianakis, K.; Tanaka, K.; Amoako, A.; et al. Altered differentiation of endometrial mesenchymal stromal fibroblasts is associated with endometriosis susceptibility. Commun. Biol. 2022, 5, 600. [Google Scholar] [CrossRef]
- Haberle, V.; Forrest, A.; Hayashizaki, Y.; Carninci, P.; Lenhard, B. CAGEr: Precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Res. 2015, 43, e51. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.; Corces, V.G. Enhancers: Emerging roles in cell fate specification. EMBO Rep. 2012, 13, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Murakawa, Y.; Yoshihara, M.; Kawaji, H.; Nishikawa, M.; Zayed, H.; Suzuki, H.; FANTOM Consortium; Hayashizaki, Y. Enhanced Identification of Transcriptional Enhancers Provides Mechanistic Insights into Diseases. Trends Genet. 2016, 32, 76–88. [Google Scholar] [CrossRef]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Thodberg, M.; Thieffry, A.; Vitting-Seerup, K.; Andersson, R.; Sandelin, A. CAGEfightR: Analysis of 5′-end data using R/Bioconductor. BMC Bioinform. 2019, 20, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Mikhaylichenko, O.; Bondarenko, V.; Harnett, D.; Schor, I.E.; Males, M.; Viales, R.R.; Furlong, E.E.M. The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev. 2018, 32, 42–57. [Google Scholar] [CrossRef] [Green Version]
- Varshney, A.; Kyono, Y.; Elangovan, V.R.; Wang, C.; Erdos, M.R.; Narisu, N.; Albanus, R.D.; Orchard, P.; Stitzel, M.L.; Collins, F.S.; et al. A Transcription Start Site Map in Human Pancreatic Islets Reveals Functional Regulatory Signatures. Diabetes 2021, 70, 1581–1591. [Google Scholar] [CrossRef]
- Kawaji, H.; Lizio, M.; Itoh, M.; Kanamori-Katayama, M.; Kaiho, A.; Nishiyori-Sueki, H.; Shin, J.W.; Kojima-Ishiyama, M.; Kawano, M.; Murata, M.; et al. Comparison of CAGE and RNA-seq transcriptome profiling using clonally amplified and single-molecule next-generation sequencing. Genome Res. 2014, 24, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Meola, J.; Hidalgo, G.D.S.; e Silva, J.C.R.; Silva, L.E.C.M.; Paz, C.C.P.; Ferriani, R.A. Caldesmon: New Insights for Diagnosing Endometriosis1. Biol. Reprod. 2013, 88, 122. [Google Scholar] [CrossRef]
- Huang, M.; Li, X.; Guo, P.; Yu, Z.; Xu, Y.; Wei, Z. The abnormal expression of oxytocin receptors in the uterine junctional zone in women with endometriosis. Reprod. Biol. Endocrinol. 2017, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Dsupin, B.A.; Guidice, L.C.; Bondy, C.A. Insulin-like growth factor system gene expression in human endometrium during the menstrual cycle. J. Clin. Endocrinol. Metab. 1994, 79, 1723–1734. [Google Scholar] [PubMed] [Green Version]
- Hu, W.-P.; Tay, S.K.; Zhao, Y. Endometriosis-Specific Genes Identified by Real-Time Reverse Transcription-Polymerase Chain Reaction Expression Profiling of Endometriosis Versus Autologous Uterine Endometrium. J. Clin. Endocrinol. Metab. 2006, 91, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, C.; Allard, J.B. Insulin-Like Growth Factor Binding Protein-5 in Physiology and Disease. Front. Endocrinol. 2020, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Gusev, N.B. Some properties of caldesmon and calponin and the participation of these proteins in regulation of smooth muscle contraction and cytoskeleton formation. Biochemistry 2001, 66, 1112–1121. [Google Scholar] [CrossRef]
- Meola, J.; Rosa e Silva, J.C.; Dentillo, D.B.; da Silva, W.A., Jr.; Veiga-Castelli, L.C.; Bernardes, L.A.; Ferriani, R.A.; de Paz, C.C.; Giuliatti, S.; Martelli, L. Differentially expressed genes in eutopic and ectopic endometrium of women with endometriosis. Fertil. Steril. 2010, 93, 1750–1773. [Google Scholar] [CrossRef]
- Kim, K.-H.; Yeo, S.-G.; Kim, W.K.; Kim, D.Y.; Yeo, H.Y.; Hong, J.P.; Chang, H.J.; Park, J.W.; Kim, S.Y.; Kim, B.C.; et al. Up-regulated expression of l-caldesmon associated with malignancy of colorectal cancer. BMC Cancer 2012, 12, 601. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-P.; Wang, C.-L.A.; Kao, H.-K.; Liang, Y.; Bs, S.-C.L.; Bs, L.-L.H.; Hseuh, C.; Hsieh, Y.-J.; Chien, K.-Y.; Chang, Y.-S.; et al. Overexpression of caldesmon is associated with lymph node metastasis and poorer prognosis in patients with oral cavity squamous cell carcinoma. Cancer 2013, 119, 4003–4011. [Google Scholar] [CrossRef]
- Lee, M.-S.; Lee, J.; Kim, J.H.; Kim, W.T.; Kim, W.-J.; Ahn, H.; Park, J. Overexpression of caldesmon is associated with tumor progression in patients with primary non-muscle-invasive bladder cancer. Oncotarget 2015, 6, 40370–40384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, H.; Wang, A.; Shen, Y.; Wang, Q.; Zhou, Z.; Zhang, R.; Li, K.; Liu, C.; Jia, H. Identification of novel alternative splicing isoform biomarkers and their association with overall survival in colorectal cancer. BMC Gastroenterol. 2020, 20, 171. [Google Scholar] [CrossRef] [PubMed]
- Vouk, K.; Šmuc, T.; Guggenberger, C.; Ribič-Pucelj, M.; Šinkovec, J.; Husen, B.; Thole, H.; Houba, P.; Thaete, C.; Adamski, J.; et al. Novel estrogen-related genes and potential biomarkers of ovarian endometriosis identified by differential expression analysis. J. Steroid Biochem. Mol. Biol. 2011, 125, 231–242. [Google Scholar] [CrossRef]
- Mettler, L.; Salmassi, A.; Schollmeyer, T.; Schmutzler, A.G.; Püngel, F.; Jonat, W. Comparison of c-DNA microarray analysis of gene expression between eutopic endometrium and ectopic endometrium (endometriosis). J. Assist. Reprod. Genet. 2007, 24, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojo, H.; Kaneko, Y. Characterization and Expression of Oxytocin and the Oxytocin Receptor. Mol. Genet. Metab. 2000, 71, 552–558. [Google Scholar] [CrossRef]
- Mechsner, S.; Bartley, J.; Loddenkemper, C.; Salomon, D.S.; Starzinski-Powitz, A.; Ebert, A.D. Oxytocin receptor expression in smooth muscle cells of peritoneal endometriotic lesions and ovarian endometriotic cysts. Fertil. Steril. 2005, 83 (Suppl. 4), 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.-R.; Fuchs, F.; Soloff, M.S. Oxytocin Receptors in Nonpregnant Human Uterus. J. Clin. Endocrinol. Metab. 1985, 60, 37–41. [Google Scholar] [CrossRef]
- Guo, S.-W.; Mao, X.; Ma, Q.; Liu, X. Dysmenorrhea and its severity are associated with increased uterine contractility and overexpression of oxytocin receptor (OTR) in women with symptomatic adenomyosis. Fertil. Steril. 2013, 99, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Li, L.; Shui, S.; Yang, L.; Sui, C.; Zhang, H. Aberrant expression of oxytocin receptor in endometrium and decidua in women who have experienced recurrent implantation failure. F&S Sci. 2020, 1, 183–187. [Google Scholar] [CrossRef]
- Stranger, E.B.; Nica, A.C.; Forrest, M.S.; Dimas, A.; Bird, C.P.; Beazley, C.; Ingle, E.C.; Dunning, M.; Flicek, P.; Koller, D.; et al. Population genomics of human gene expression. Nat. Genet. 2007, 39, 1217–1224. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Jing, C.; Li, L.; Zhang, L.; Shi, Y.; Wang, J.; Liu, J.; Li, C. Down-regulation of VEZT gene expression in human gastric cancer involves promoter methylation and miR-43c. Biochem. Biophys. Res. Commun. 2011, 404, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth-Carson, S.J.; Fung, J.N.; Luong, H.T.; Sapkota, Y.; Bowdler, L.M.; Wallace, L.; Teh, W.T.; Powell, J.E.; Girling, J.E.; Healey, M.; et al. Endometrial vezatin and its association with endometriosis risk. Hum. Reprod. 2016, 31, 999–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, I.M.; Mortlock, S.; Montgomery, G.W. Genetic Regulation of Physiological Reproductive Lifespan and Female Fertility. Int. J. Mol. Sci. 2021, 22, 2556. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. Erratum: Genetic effects on gene expression across human tissues. Nature 2018, 553, 530. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.; Hoorn, T.V.D.; Jongsma, M.L.; Bakker, M.J.; Hengeveld, R.; Janssen, L.; Cresswell, P.; Egan, D.A.; van Ham, M.; Brinke, A.T.; et al. A Genome-wide Multidimensional RNAi Screen Reveals Pathways Controlling MHC Class II Antigen Presentation. Cell 2011, 145, 268–283. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, C.M.; de Oliveira, R.; Christofolini, D.M.; Barbosa, C.P.; Bianco, B. Effects of a Polymorphism in the Promoter Region of the Follicle-Stimulating Hormone Subunit Beta (FSHB) Gene on Female Reproductive Outcomes. Genet. Test. Mol. Biomarkers 2018, 23, 39–44. [Google Scholar] [CrossRef]
- Powell, J.E.; Fung, J.N.; Shakhbazov, K.; Sapkota, Y.; Cloonan, N.; Hemani, G.; Hillman, K.M.; Kaufmann, S.; Luong, H.T.; Bowdler, L.; et al. Endometriosis risk alleles at 1p36.12 act through inverse regulation of CDC42 and LINC00339. Hum. Mol. Genet. 2016, 25, 5046–5058. [Google Scholar]
- Stengel, K.; Zheng, Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell. Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Stamatiades, G.A.; Kaiser, U.B. Gonadotropin regulation by pulsatile GnRH: Signaling and gene expression. Mol. Cell. Endocrinol. 2018, 463, 131–141. [Google Scholar] [CrossRef]
- Mei, Q.; Li, H.; Liu, Y.; Wang, X.; Xiang, W. Advances in the study of CDC42 in the female reproductive system. J. Cell. Mol. Med. 2022, 26, 16–24. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Case/Control | rAFS % | ENZIAN & | Age | BMI | Smoking # | Infertility * |
|---|---|---|---|---|---|---|---|
| Sample 1 | Case | I | 0 | 43 | 25 | 0 | 0 |
| Sample 2 | Control | 0 | 0 | 51 | 24.3 | 0 | 0 |
| Sample 3 | Control | 0 | 0 | 41 | 24.2 | 0 | 1a |
| Sample 4 | Case | I | A0B2C0 | 46 | 20.7 | 0 | 0 |
| Sample 5 | Case | I | 0 | 36 | 22.9 | 1 | 1a |
| Sample 6 | Case | II | 2B | 31 | 21.3 | 0 | 0 |
| Sample 7 | Control | 0 | 0 | 25.7 | 28.7 | n.d. | 0 |
| Sample 8 | Control | 0 | 0 | 21.7 | 17.2 | 0 | 0 |
| Sample ID | Case/Control | Commercial | In-House |
|---|---|---|---|
| Sample 1 | Case | 13,878,668 | 7,424,199 |
| Sample 2 | Control | 11,976,955 | 6,373,486 |
| Sample 3 | Control | 10,337,586 | 6,380,118 |
| Sample 4 | Case | 13,202,981 | 6,579,239 |
| Sample 5 | Case | 14,025,009 | 5,394,546 |
| Sample 6 | Case | 14,100,380 | 7,617,671 |
| Sample 7 | Control | 16,107,917 | 9,768,981 |
| Sample 8 | Control | 20,367,598 | 7,825,180 |
| Sample_ID | Case/Control | Number of Tag Clusters | ATAC-Seq Peaks | No. of Tag Clusters Overlapping with ATAC-Seq |
|---|---|---|---|---|
| Sample 1 | Case | 8343 | 114583 | 6688 |
| Sample 2 | Control | 8337 | 138093 | 6933 |
| Sample 3 | Control | 8327 | 125615 | 6869 |
| Sample 4 | Case | 8346 | 116957 | 6860 |
| Sample 5 | Case | 8319 | 137215 | 7005 |
| Sample 6 | Case | 8346 | 121518 | 6913 |
| Sample 7 | Control | 8389 | 122149 | 6834 |
| Sample 8 | Control | 8403 | 122214 | 6802 |
| Sample ID | Case/Control | Tag Clusters |
|---|---|---|
| Sample 1 | Case | 8343 |
| Sample 2 | Control | 8337 |
| Sample 3 | Control | 8327 |
| Sample 4 | Case | 8346 |
| Sample 5 | Case | 8319 |
| Sample 6 | Case | 8346 |
| Sample 7 | Control | 8389 |
| Sample 8 | Control | 8403 |
| Chr | Start | End | Strand | TPM # | Annotation | Genes | Log2FC | p-Value | FDR * |
|---|---|---|---|---|---|---|---|---|---|
| chr2 | 216694838 | 216694915 | − | 13.00 | promoter | IGFBP5 | 3.05 | 3.00 × 10−16 | 2.13 × 10−12 |
| chr3 | 8769610 | 8769628 | − | 26.97 | promoter | OXTR | 2.65 | 2.24 × 10−15 | 7.93 × 10−12 |
| chr2 | 216695547 | 216695559 | − | 1476.3 | promoter | IGFBP5 | 2.59 | 5.23 × 10−14 | 1.24 × 10−10 |
| chr5 | 147906536 | 147906541 | − | 10.87 | promoter | C5orf46 | 3.62 | 1.29 × 10−12 | 2.29 × 10−9 |
| chr14 | 101964571 | 101964575 | + | 44.23 | promoter | DYNC1H1 | 1.00 | 6.75 × 10−12 | 9.57 × 10−9 |
| chr5 | 98773661 | 98773701 | + | 41.68 | promoter | RGMB | 1.35 | 1.23 × 10−10 | 1.45 × 10−7 |
| chr19 | 43205633 | 43205648 | − | 108.38 | promoter | PSG4 | 3.74 | 1.89 × 10−10 | 1.92 × 10−7 |
| chr4 | 186726665 | 186726728 | − | 28.46 | promoter | FAT1 | 1.58 | 2.54 × 10−10 | 2.25 × 10−7 |
| chr2 | 216676450 | 216676709 | − | 80.13 | exon | IGFBP5 | 2.47 | 3.49 × 10−10 | 2.75 × 10−7 |
| chr4 | 114364911 | 114364951 | − | 27.86 | unknown | 2.37 | 8.15 × 10−10 | 5.78 × 10−7 | |
| chr2 | 216674321 | 216674452 | − | 20.13 | exon | IGFBP5 | 2.76 | 1.67 × 10−9 | 1.08 × 10−6 |
| chr1 | 218345334 | 218345346 | + | 76.83 | promoter | TGFB2 | 2.44 | 3.02 × 10−9 | 1.78 × 10−6 |
| chr7 | 134711476 | 134711515 | + | 34.95 | unknown | 1.45 | 3.99 × 10−9 | 2.17 × 10−6 | |
| chr9 | 78297122 | 78297161 | + | 36.46 | promoter | PSAT1 | −1.94 | 6.68 × 10−9 | 3.38 × 10−6 |
| chr3 | 188212669 | 188212713 | + | 37.34 | promoter | LPP | 1.858 | 1.19 × 10−8 | 5.62 × 10−6 |
| chr7 | 134646835 | 134646861 | + | 29.33 | promoter | BPGM | 0.95 | 1.91 × 10−8 | 8.47 × 10−6 |
| chr2 | 216695059 | 216695140 | − | 8.79 | promoter | IGFBP5 | 2.65 | 3.88 × 10−8 | 1.62 × 10−5 |
| chr4 | 94451901 | 94451973 | + | 109.58 | promoter | PDLIM5 | 0.83 | 5.87 × 10−8 | 2.31 × 10−5 |
| chr6 | 26021572 | 26021654 | + | 23.35 | promoter | −1.43 | 6.45 × 10−8 | 2.40 × 10−5 | |
| chr5 | 40679914 | 40679919 | + | 5.58 | promoter | PTGER4 | −1.22 | 7.21 × 10−8 | 2.55 × 10−5 |
| chr16 | 3065631 | 3065641 | + | 6.75 | promoter | IL32 | 2.33 | 9.46 × 10−8 | 3.19 × 10−5 |
| chr9 | 72953039 | 72953073 | − | 30.88 | promoter | ALDH1A1 | −2.26 | 2.21 × 10−7 | 7.13 × 10−5 |
| chr6 | 131949556 | 131949569 | − | 4.73 | exon | CCN2 | 1.85 | 2.71 × 10−7 | 8.33 × 10−5 |
| chr17 | 80260832 | 80260882 | + | 19.59 | promoter | AC124319.1 | 1.02 | 2.94 × 10−7 | 8.69 × 10−5 |
| chr12 | 56315875 | 56316039 | − | 86.96 | promoter | AC073896.1; CNPY2 | -0.44 | 3.73 × 10−7 | 0.00010 |
| chr18 | 45967267 | 45967330 | − | 21.44 | promoter | EPG5 | 0.838 | 4.59 × 10−7 | 0.00012 |
| chr2 | 216675681 | 216675683 | − | 2.52 | exon | IGFBP5 | 2.85 | 4.70 × 10−7 | 0.00012 |
| chr15 | 63042680 | 63042756 | + | 659.91 | promoter | TPM1 | 0.86 | 4.96 × 10−7 | 0.00012 |
| chr2 | 216676128 | 216676131 | − | 3.19 | exon | IGFBP5 | 3.59 | 5.28 × 10−7 | 0.00012 |
| chr7 | 134867737 | 134867770 | + | 7.57 | exon | CALD1 | 1.49 | 6.20 × 10−7 | 0.00014 |
| chr5 | 178204530 | 178204537 | + | 114.22 | promoter | HNRNPAB | −0.75 | 7.73 × 10−7 | 0.00017 |
| chr9 | 38392671 | 38392799 | + | 44.95 | promoter | ALDH1B1 | 1.954 | 9.92 × 10−7 | 0.00021 |
| chr9 | 5510498 | 5510556 | + | 16.50 | promoter | PDCD1LG2 | 1.149 | 1.25 × 10−6 | 0.00026 |
| chr3 | 156674590 | 156674634 | + | 27.80 | promoter | TIPARP | 0.93 | 1.29 × 10−6 | 0.00026 |
| chr8 | 23404118 | 23404156 | − | 732.82 | promoter | LOXL2; ENTPD4 | 0.823 | 1.43 × 10−6 | 0.00028 |
| chr12 | 29783910 | 29783922 | − | 17.55 | promoter | TMTC1 | −1.76 | 1.55 × 10−6 | 0.00029 |
| chr8 | 41309471 | 41309474 | − | 3.24 | promoter | SFRP1 | −2.64 | 1.53 × 10−6 | 0.00029 |
| chr5 | 139293674 | 139293754 | + | 75.81 | promoter | MATR3 | -0.53 | 1.61 × 10−6 | 0.00029 |
| chr5 | 84384380 | 84384483 | − | 19.82 | promoter | EDIL3 | 1.62 | 1.89 × 10−6 | 0.00034 |
| chr20 | 50190829 | 50190835 | + | 51.41 | promoter | CEBPB | −0.855 | 2.31 × 10−6 | 0.00040 |
| chr11 | 62546749 | 62546845 | − | 101.41 | promoter | AHNAK | 1.031 | 2.43 × 10−6 | 0.00041 |
| chr7 | 134928752 | 134928863 | + | 21.03 | exon | CALD1 | 0.99 | 2.80 × 10−6 | 0.00047 |
| chr11 | 117204261 | 117204391 | + | 42.70 | exon | TAGLN | 0.93 | 2.98 × 10−6 | 0.00047 |
| chr5 | 141969105 | 141969137 | + | 6.875 | promoter | RNF14 | 1.66 | 2.95 × 10−6 | 0.00047 |
| chr10 | 32957884 | 32957980 | − | 58.70 | promoter | ITGB1 | 1.192 | 3.16 × 10−6 | 0.00049 |
| chr20 | 63696646 | 63696657 | + | 16.99 | promoter | RTEL1-TNFRSF6B; TNFRSF6B | 1.405 | 3.96 × 10−6 | 0.00060 |
| chr1 | 109687817 | 109687847 | + | 4.17 | promoter | GSTM2; GSTM1 | 5.476 | 4.47 × 10−6 | 0.00067 |
| chr16 | 71358723 | 71358731 | + | 93.20 | promoter | CALB2 | 2.51 | 4.96 × 10−6 | 0.00073 |
| chr2 | 30231709 | 30231716 | + | 7.77 | promoter | LBH | 1.24 | 5.44 × 10−6 | 0.00078 |
| chr2 | 216695357 | 216695370 | − | 52.35 | promoter | IGFBP5 | 2.43 | 5.64 × 10−6 | 0.00079 |
| chr2 | 216372053 | 216372078 | − | 6.62 | promoter | MARCHF4 | 1.08 | 5.83 × 10−6 | 0.00080 |
| chr1 | 78004920 | 78004954 | + | 30.40 | promoter | DNAJB4; GIPC2 | 1.31 | 5.90 × 10−6 | 0.00080 |
| chr9 | 116153791 | 116153813 | + | 44.29214 | promoter | PAPPA | 1.59 | 6.49 × 10−6 | 0.00086 |
| Chr | BP | SNP | p-Value | Allele | Gene Region |
|---|---|---|---|---|---|
| chr1 | 22051787 | rs725158 | 4.88 × 10−16 | a | CDC42 |
| chr1 | 22052387 | rs3754496 | 4.99 × 10−16 | a | CDC42 |
| chr2 | 215433073 | rs1250244 | 8.93 × 10−8 | c | FN1 |
| chr7 | 137345599 | rs161335 | 4.67 × 10−6 | t | next to PTN |
| chr11 | 30322210 | rs3858429 | 5.45 × 10−8 | t | XR_931152.2 |
| chr11 | 30323044 | rs4071558 | 5.62 × 10−8 | t | near ARL14EP |
| chr11 | 30323178 | rs4071559 | 5.60 × 10−8 | t | ARL14EP |
| chr12 | 95216444 | rs7310833 | 8.07 × 10−9 | a | FGD6 |
| chr12 | 95217365 | rs6538617 | 3.60 × 10−6 | t | FGD6 |
| chr12 | 95217409 | rs6538618 | 6.68 × 10−9 | t | FGD6 |
| chr17 | 44898335 | rs35653192 | 9.15 × 10−6 | a | EFTUD2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marla, S.; Mortlock, S.; Yoon, S.; Crawford, J.; Andersen, S.; Mueller, M.D.; McKinnon, B.; Nguyen, Q.; Montgomery, G.W. Global Analysis of Transcription Start Sites and Enhancers in Endometrial Stromal Cells and Differences Associated with Endometriosis. Cells 2023, 12, 1736. https://doi.org/10.3390/cells12131736
Marla S, Mortlock S, Yoon S, Crawford J, Andersen S, Mueller MD, McKinnon B, Nguyen Q, Montgomery GW. Global Analysis of Transcription Start Sites and Enhancers in Endometrial Stromal Cells and Differences Associated with Endometriosis. Cells. 2023; 12(13):1736. https://doi.org/10.3390/cells12131736
Chicago/Turabian StyleMarla, Sushma, Sally Mortlock, Sohye Yoon, Joanna Crawford, Stacey Andersen, Michael D. Mueller, Brett McKinnon, Quan Nguyen, and Grant W. Montgomery. 2023. "Global Analysis of Transcription Start Sites and Enhancers in Endometrial Stromal Cells and Differences Associated with Endometriosis" Cells 12, no. 13: 1736. https://doi.org/10.3390/cells12131736
APA StyleMarla, S., Mortlock, S., Yoon, S., Crawford, J., Andersen, S., Mueller, M. D., McKinnon, B., Nguyen, Q., & Montgomery, G. W. (2023). Global Analysis of Transcription Start Sites and Enhancers in Endometrial Stromal Cells and Differences Associated with Endometriosis. Cells, 12(13), 1736. https://doi.org/10.3390/cells12131736