

Emerging Role of Kinin B1 Receptor in Persistent Neuroinflammation and Neuropsychiatric Symptoms in Mice Following Recovery from SARS-CoV-2 Infection

 ,
,  , ,
, ,
Abstract
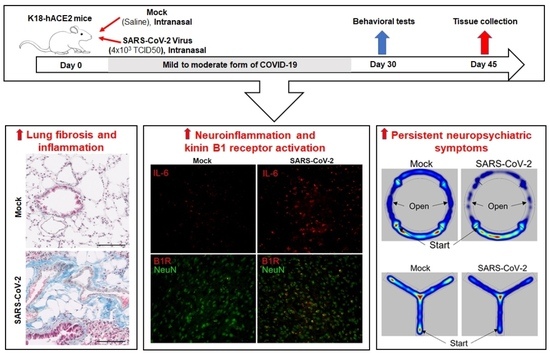
:
1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. SARS-CoV-2 Virus
2.3. Experimental Design
2.4. Behavior and Cognitive Function Tests
2.5. Histology and Immunohistochemistry
2.6. RNA Extraction and Real-Time Quantitative RT-PCR
2.7. SARS-CoV-2 Antibody Titer Measurement Using ELISA
2.8. Statistical Analysis
3. Results
3.1. K18-hACE2 Mice Are Susceptible to Severe SARS-CoV-2 Infection
3.2. Severe SARS-CoV-2 Infection Increases Inflammatory Markers in the Brain
3.3. Mild SARS-CoV-2 Infection Causes Significant Antibody Generation
3.4. Mild SARS-CoV-2 Infection Causes Lung Fibrosis and Increased B1R Expression
3.5. Mild SARS-CoV-2 Infection Increases Inflammatory Markers in the Brain
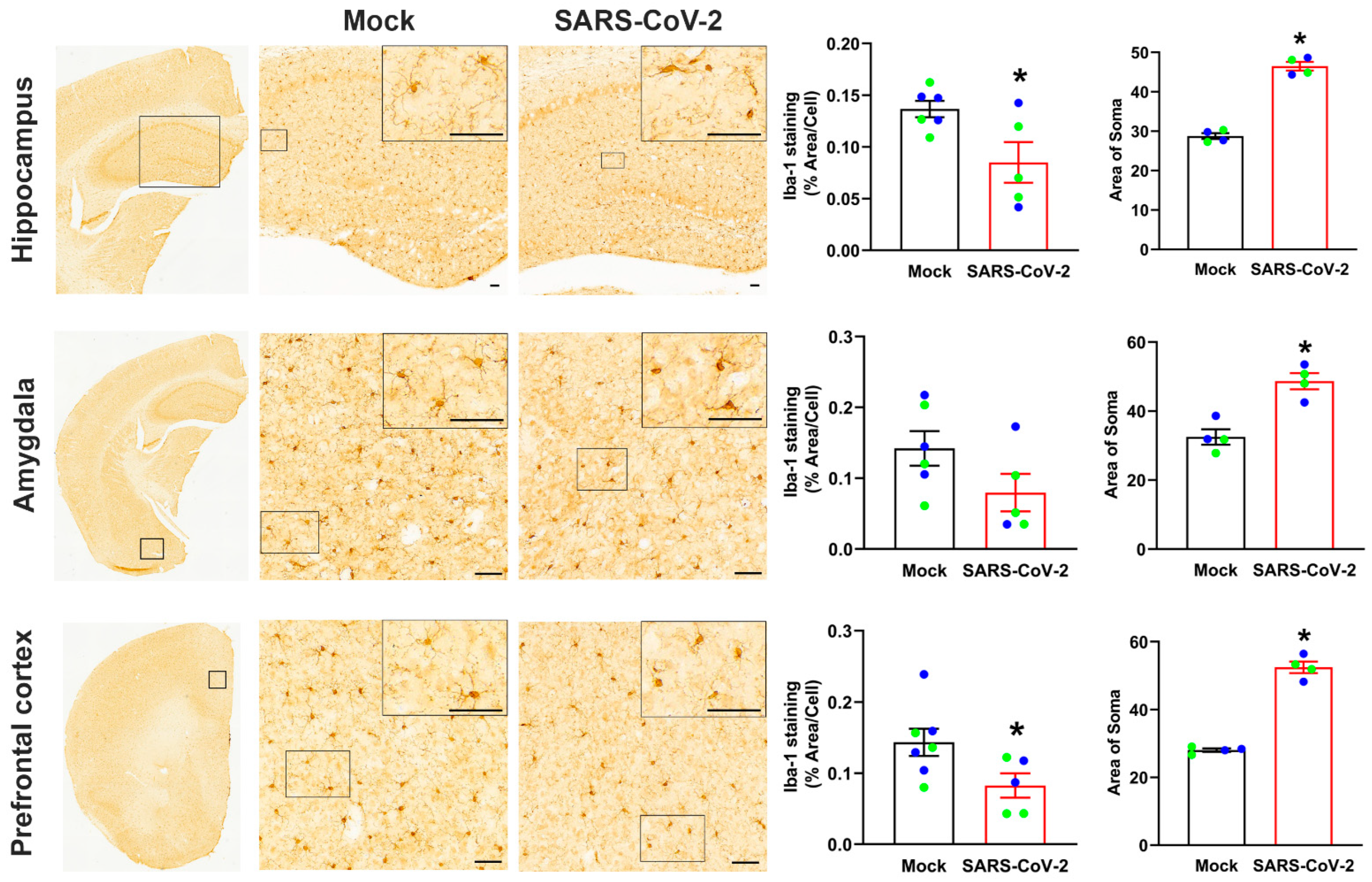
3.6. Mild SARS-CoV-2 Infection Results in Persistent Activation of Microglia in the Brain
3.7. Mild SARS-CoV-2 Infection Causes Neuropsychiatric and Behavioral Symptoms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larsen, J.R.; Martin, M.R.; Martin, J.D.; Kuhn, P.; Hicks, J.B. Modeling the Onset of Symptoms of COVID-19. Front. Public Health 2020, 8, 473. [Google Scholar] [CrossRef]
- Buszko, M.; Park, J.H.; Verthelyi, D.; Sen, R.; Young, H.A.; Rosenberg, A.S. The dynamic changes in cytokine responses in COVID-19: A snapshot of the current state of knowledge. Nat. Immunol. 2020, 21, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.A.; Roche, R. A hypothesized role for dysregulated bradykinin signaling in COVID-19 respiratory complications. FASEB J. 2020, 34, 7265–7269. [Google Scholar] [CrossRef] [PubMed]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.K.; Franko, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef]
- Taquet, M.; Geddes, J.R.; Husain, M.; Luciano, S.; Harrison, P.J. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: A retrospective cohort study using electronic health records. Lancet Psychiatry 2021, 8, 416–427. [Google Scholar] [CrossRef]
- Graham, E.L.; Clark, J.R.; Orban, Z.S.; Lim, P.H.; Szymanski, A.L.; Taylor, C.; DiBiase, R.M.; Jia, D.T.; Balabanov, R.; Ho, S.U.; et al. Persistent neurologic symptoms and cognitive dysfunction in non-hospitalized Covid-19 "long haulers". Ann. Clin. Transl. Neurol. 2021, 8, 1073–1085. [Google Scholar] [CrossRef]
- Reiken, S.; Sittenfeld, L.; Dridi, H.; Liu, Y.; Liu, X.; Marks, A.R. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimers Dement. 2022, 18, 955–965. [Google Scholar] [CrossRef]
- Cocoros, N.M.; Svensson, E.; Szepligeti, S.K.; Vestergaard, S.V.; Szentkuti, P.; Thomsen, R.W.; Borghammer, P.; Sorensen, H.T.; Henderson, V.W. Long-term Risk of Parkinson Disease Following Influenza and Other Infections. JAMA Neurol. 2021, 78, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Vigasova, D.; Nemergut, M.; Liskova, B.; Damborsky, J. Multi-pathogen infections and Alzheimer’s disease. Microb Cell Fact. 2021, 20, 25. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, R.E.; Martynova, E.; Rizvanov, A.A.; Khaiboullina, S.; Verma, S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses 2020, 12, 643. [Google Scholar] [CrossRef] [PubMed]
- Guedj, E.; Million, M.; Dudouet, P.; Tissot-Dupont, H.; Bregeon, F.; Cammilleri, S.; Raoult, D. (18)F-FDG brain PET hypometabolism in post-SARS-CoV-2 infection: Substrate for persistent/delayed disorders? Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.J. Long COVID or post-COVID-19 syndrome: Putative pathophysiology, risk factors, and treatments. Infect Dis. 2021, 53, 737–754. [Google Scholar] [CrossRef]
- Liu, J.M.; Tan, B.H.; Wu, S.; Gui, Y.; Suo, J.L.; Li, Y.C. Evidence of central nervous system infection and neuroinvasive routes, as well as neurological involvement, in the lethality of SARS-CoV-2 infection. J. Med. Virol. 2021, 93, 1304–1313. [Google Scholar] [CrossRef]
- Lewis, A.; Frontera, J.; Placantonakis, D.G.; Lighter, J.; Galetta, S.; Balcer, L.; Melmed, K.R. Cerebrospinal fluid in COVID-19: A systematic review of the literature. J. Neurol. Sci. 2021, 421, 117316. [Google Scholar] [CrossRef]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612, 758–763. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Savitt, A.G.; Manimala, S.; White, T.; Fandaros, M.; Yin, W.; Duan, H.; Xu, X.; Geisbrecht, B.V.; Rubenstein, D.A.; Kaplan, A.P.; et al. SARS-CoV-2 Exacerbates COVID-19 Pathology Through Activation of the Complement and Kinin Systems. Front. Immunol. 2021, 12, 767347. [Google Scholar] [CrossRef]
- Jia, H.; Yue, X.; Lazartigues, E. ACE2 mouse models: A toolbox for cardiovascular and pulmonary research. Nat. Commun. 2020, 11, 5165. [Google Scholar] [CrossRef]
- Dinnon, K.H., 3rd; Leist, S.R.; Schafer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L., Jr.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 2020, 586, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 583, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Port, J.R.; Bushmaker, T.; Offei Owusu, I.; Purushotham, J.N.; Avanzato, V.A.; Fischer, R.J.; Schulz, J.E.; Holbrook, M.G.; Hebner, M.J.; et al. K18-hACE2 mice develop respiratory disease resembling severe COVID-19. PLoS Pathog. 2021, 17, e1009195. [Google Scholar] [CrossRef]
- Sun, S.H.; Chen, Q.; Gu, H.J.; Yang, G.; Wang, Y.X.; Huang, X.Y.; Liu, S.S.; Zhang, N.N.; Li, X.F.; Xiong, R.; et al. A Mouse Model of SARS-CoV-2 Infection and Pathogenesis. Cell Host Microbe 2020, 28, 124–133 e124. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Mead, H.; Tian, L.; Park, J.G.; Garcia, J.I.; Jaramillo, S.; Barr, T.; Kollath, D.S.; Coyne, V.K.; Stone, N.E.; et al. The K18-Human ACE2 Transgenic Mouse Model Recapitulates Non-severe and Severe COVID-19 in Response to an Infectious Dose of the SARS-CoV-2 Virus. J. Virol. 2022, 96, e0096421. [Google Scholar] [CrossRef]
- Carossino, M.; Kenney, D.; O’Connell, A.K.; Montanaro, P.; Tseng, A.E.; Gertje, H.P.; Grosz, K.A.; Ericsson, M.; Huber, B.R.; Kurnick, S.A.; et al. Fatal Neurodissemination and SARS-CoV-2 Tropism in K18-hACE2 Mice Is Only Partially Dependent on hACE2 Expression. Viruses 2022, 14, 535. [Google Scholar] [CrossRef]
- Parekh, R.U.; Robidoux, J.; Sriramula, S. Kinin B1 Receptor Blockade Prevents Angiotensin II-induced Neuroinflammation and Oxidative Stress in Primary Hypothalamic Neurons. Cell Mol. Neurobiol. 2020, 40, 845–857. [Google Scholar] [CrossRef]
- Akula, S.M.; Bolin, P.; Cook, P.P. Cellular miR-150-5p may have a crucial role to play in the biology of SARS-CoV-2 infection by regulating nsp10 gene. RNA Biol. 2022, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Morrison, H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018, h. [Google Scholar] [CrossRef]
- Morrison, H.; Young, K.; Qureshi, M.; Rowe, R.K.; Lifshitz, J. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep. 2017, 7, 13211. [Google Scholar] [CrossRef]
- Parekh, R.U.; Sriramula, S. Activation of Kinin B1R Upregulates ADAM17 and Results in ACE2 Shedding in Neurons. Int. J. Mol. Sci. 2020, 22, 145. [Google Scholar] [CrossRef]
- Sriramula, S.; Lazartigues, E. Kinin B1 Receptor Promotes Neurogenic Hypertension Through Activation of Centrally Mediated Mechanisms. Hypertension 2017, 70, 1122–1131. [Google Scholar] [CrossRef]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef]
- John, A.E.; Joseph, C.; Jenkins, G.; Tatler, A.L. COVID-19 and pulmonary fibrosis: A potential role for lung epithelial cells and fibroblasts. Immunol. Rev. 2021, 302, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Leon-Rodriguez, A.; Fernandez-Arjona, M.D.M.; Grondona, J.M.; Pedraza, C.; Lopez-Avalos, M.D. Anxiety-like behavior and microglial activation in the amygdala after acute neuroinflammation induced by microbial neuraminidase. Sci. Rep. 2022, 12, 11581. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.W.; Cline, C.R.; Zeng, X.; Garrison, A.R.; Carey, B.D.; Mucker, E.M.; White, L.E.; Shamblin, J.D.; Brocato, R.L.; Liu, J.; et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Oladunni, F.S.; Park, J.G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allue-Guardia, A.; Olmo-Fontanez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Castaneda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.H.; Wood, J.; Yalcin, B.; Taylor, K.R.; Dutton, S.; Acosta-Alvarez, L.; et al. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. bioRxiv 2022. [CrossRef]
- Kumari, P.; Rothan, H.A.; Natekar, J.P.; Stone, S.; Pathak, H.; Strate, P.G.; Arora, K.; Brinton, M.A.; Kumar, M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses 2021, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Soung, A.; Sissoko, C.; Nordvig, A.; Canoll, P.; Mariani, M.; Jiang, X.; Bricker, T.; Goldman, J.; Rosoklija, G.; et al. COVID-19 induces neuroinflammation and loss of hippocampal neurogenesis. Res. Sq. 2021. [CrossRef]
- Philippens, I.; Boszormenyi, K.P.; Wubben, J.A.M.; Fagrouch, Z.C.; van Driel, N.; Mayenburg, A.Q.; Lozovagia, D.; Roos, E.; Schurink, B.; Bugiani, M.; et al. Brain Inflammation and Intracellular alpha-Synuclein Aggregates in Macaques after SARS-CoV-2 Infection. Viruses 2022, 14, 776. [Google Scholar] [CrossRef] [PubMed]
- Rutkai, I.; Mayer, M.G.; Hellmers, L.M.; Ning, B.; Huang, Z.; Monjure, C.J.; Coyne, C.; Silvestri, R.; Golden, N.; Hensley, K.; et al. Neuropathology and virus in brain of SARS-CoV-2 infected non-human primates. Nat. Commun. 2022, 13, 1745. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, W.; Chen, F.; Cui, L. COVID-19 and cognitive impairment: Neuroinvasive and blood—brain barrier dysfunction. J. Neuroinflamm. 2022, 19, 222. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Woodley, C.; Chao, L.; Margolius, H.S. Identification of tissue kallikrein in brain and in the cell-free translation product encoded by brain mRNA. J. Biol. Chem. 1983, 258, 15173–15178. [Google Scholar] [CrossRef] [PubMed]
- Albert-Weissenberger, C.; Stetter, C.; Meuth, S.G.; Gobel, K.; Bader, M.; Siren, A.L.; Kleinschnitz, C. Blocking of bradykinin receptor B1 protects from focal closed head injury in mice by reducing axonal damage and astroglia activation. J. Cereb. Blood Flow Metab. 2012, 32, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Raslan, F.; Schwarz, T.; Meuth, S.G.; Austinat, M.; Bader, M.; Renne, T.; Roosen, K.; Stoll, G.; Siren, A.L.; Kleinschnitz, C. Inhibition of bradykinin receptor B1 protects mice from focal brain injury by reducing blood-brain barrier leakage and inflammation. J. Cereb. Blood Flow Metab. 2010, 30, 1477–1486. [Google Scholar] [CrossRef]
- Trabold, R.; Eros, C.; Zweckberger, K.; Relton, J.; Beck, H.; Nussberger, J.; Muller-Esterl, W.; Bader, M.; Whalley, E.; Plesnila, N. The role of bradykinin B(1) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 2010, 30, 130–139. [Google Scholar] [CrossRef]
- Witcher, K.G.; Bray, C.E.; Chunchai, T.; Zhao, F.; O’Neil, S.M.; Gordillo, A.J.; Campbell, W.A.; McKim, D.B.; Liu, X.; Dziabis, J.E.; et al. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J. Neurosci. 2021, 41, 1597–1616. [Google Scholar] [CrossRef]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef]
- Cunningham, C.; Campion, S.; Teeling, J.; Felton, L.; Perry, V.H. The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C). Brain Behav. Immun. 2007, 21, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, B.; Al-Doaiss, A.; Shati, A.; Al-Kahtani, M.; Jarrar, Q. Behavioural alterations induced by chronic exposure to 10 nm silicon dioxide nanoparticles. IET Nanobiotechnol. 2021, 15, 221–235. [Google Scholar] [CrossRef]
- Li, W.; Chen, M.; Feng, X.; Song, M.; Shao, M.; Yang, Y.; Zhang, L.; Liu, Q.; Lv, L.; Su, X. Maternal immune activation alters adult behavior, intestinal integrity, gut microbiota and the gut inflammation. Brain Behav. 2021, 11, e02133. [Google Scholar] [CrossRef] [PubMed]
- Kitanaka, J.; Kitanaka, N.; Hall, F.S.; Fujii, M.; Goto, A.; Kanda, Y.; Koizumi, A.; Kuroiwa, H.; Mibayashi, S.; Muranishi, Y.; et al. Memory impairment and reduced exploratory behavior in mice after administration of systemic morphine. J. Exp. Neurosci. 2015, 9, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Darwish, B.; Chamaa, F.; Al-Chaer, E.D.; Saade, N.E.; Abou-Kheir, W. Intranigral Injection of Endotoxin Suppresses Proliferation of Hippocampal Progenitor Cells. Front. Neurosci. 2019, 13, 687. [Google Scholar] [CrossRef]
- Heiss, R.; Grodzki, D.M.; Horger, W.; Uder, M.; Nagel, A.M.; Bickelhaupt, S. High-performance low field MRI enables visualization of persistent pulmonary damage after COVID-19. Magn. Reson. Imaging 2021, 76, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.L.; Villacreses, R.; Nagpal, P.; Guo, J.; Pezzulo, A.A.; Thurman, A.L.; Hamzeh, N.Y.; Blount, R.J.; Fortis, S.; Hoffman, E.A.; et al. Quantitative Chest CT Assessment of Small Airways Disease in Post-Acute SARS-CoV-2 Infection. Radiology 2022, 212170. [Google Scholar] [CrossRef]
- Sollini, M.; Morbelli, S.; Ciccarelli, M.; Cecconi, M.; Aghemo, A.; Morelli, P.; Chiola, S.; Gelardi, F.; Chiti, A. Long COVID hallmarks on [18F]FDG-PET/CT: A case-control study. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3187–3197. [Google Scholar] [CrossRef]
- Benedetti, F.; Palladini, M.; Paolini, M.; Melloni, E.; Vai, B.; De Lorenzo, R.; Furlan, R.; Rovere-Querini, P.; Falini, A.; Mazza, M.G. Brain correlates of depression, post-traumatic distress, and inflammatory biomarkers in COVID-19 survivors: A multimodal magnetic resonance imaging study. Brain Behav. Immun. Health 2021, 18, 100387. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, E.; Al-Aly, Z. Risks of mental health outcomes in people with covid-19: Cohort study. BMJ 2022, 376, e068993. [Google Scholar] [CrossRef]
- Sy, M.; Kitazawa, M.; Medeiros, R.; Whitman, L.; Cheng, D.; Lane, T.E.; Laferla, F.M. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am. J. Pathol. 2011, 178, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e10076117. [Google Scholar] [CrossRef] [PubMed]
- Engler-Chiurazzi, E.B.; Russell, A.E.; Povroznik, J.M.; McDonald, K.O.; Porter, K.N.; Wang, D.S.; Hammock, J.; Billig, B.K.; Felton, C.C.; Yilmaz, A.; et al. Intermittent systemic exposure to lipopolysaccharide-induced inflammation disrupts hippocampal long-term potentiation and impairs cognition in aging male mice. Brain Behav. Immun. 2023, 108, 279–291. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mock | SARS-CoV-2 | |
|---|---|---|
| Open field | ||
| Distance (cm) | 3422.17 ± 227.39 | 3443.96 ± 178.66 |
| Velocity (cm/s) | 5.70 ± 0.38 | 5.54 ± 0.36 |
| Acceleration (cm/s2) | 669.95 ± 29.46 | 664.50 ± 35.79 |
| Time moving (s) | 348.27 ± 18.84 | 364.01± 16.43 |
| Time in border (s) | 549.43 ± 16.65 | 529.06 ± 14.17 |
| Zero Maze | ||
| Entries | 19.25 ± 1.87 | 16.33 ± 1.69 |
| Entries, whole body | 4.58 ± 1.09 | 2.33 ± 0.92 |
| Time in open, whole body (s) | 35.56 ± 7.26 | 18.24 ± 7.31 |
| Y Maze | ||
| Spontaneous alternation (%) | 65.13 ± 2.85 | 64.95 ± 4.92 |
| Immobile (s) | 9.37 ± 2.76 | 24.80 ± 10.34 |
| Velocity (cm/s) | 4.24 ± 0.15 | 3.05 ± 0.27 * |
| Distance (cm) | 1272.66 ± 43.51 | 916.3 ± 79.37 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sriramula, S.; Theobald, D.; Parekh, R.U.; Akula, S.M.; O’Rourke, D.P.; Eells, J.B. Emerging Role of Kinin B1 Receptor in Persistent Neuroinflammation and Neuropsychiatric Symptoms in Mice Following Recovery from SARS-CoV-2 Infection. Cells 2023, 12, 2107. https://doi.org/10.3390/cells12162107
Sriramula S, Theobald D, Parekh RU, Akula SM, O’Rourke DP, Eells JB. Emerging Role of Kinin B1 Receptor in Persistent Neuroinflammation and Neuropsychiatric Symptoms in Mice Following Recovery from SARS-CoV-2 Infection. Cells. 2023; 12(16):2107. https://doi.org/10.3390/cells12162107
Chicago/Turabian StyleSriramula, Srinivas, Drew Theobald, Rohan Umesh Parekh, Shaw M. Akula, Dorcas P. O’Rourke, and Jeffrey B. Eells. 2023. "Emerging Role of Kinin B1 Receptor in Persistent Neuroinflammation and Neuropsychiatric Symptoms in Mice Following Recovery from SARS-CoV-2 Infection" Cells 12, no. 16: 2107. https://doi.org/10.3390/cells12162107
APA StyleSriramula, S., Theobald, D., Parekh, R. U., Akula, S. M., O’Rourke, D. P., & Eells, J. B. (2023). Emerging Role of Kinin B1 Receptor in Persistent Neuroinflammation and Neuropsychiatric Symptoms in Mice Following Recovery from SARS-CoV-2 Infection. Cells, 12(16), 2107. https://doi.org/10.3390/cells12162107