
Disheveled-1 Interacts with Claudin-5 and Contributes to Norrin-Induced Endothelial Barrier Restoration

 , and
, and
Abstract
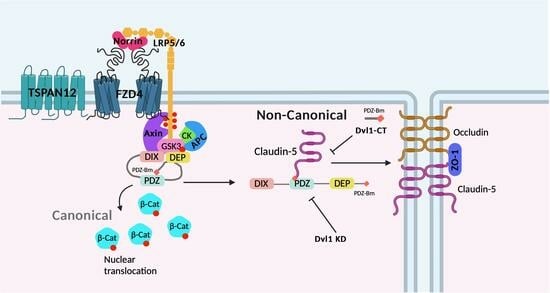
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Primary Bovine Retinal Endothelial Cell Culture (BREC)
2.2. BREC Transfection with siRNAs or DVL Mutants
2.3. HEK-293 Transfection
2.4. Co-Immunoprecipitation
2.5. Western Blot (WB)
2.6. Immunofluorescence Staining
2.7. qRT-PCR
2.8. Statistical Analysis
3. Results
3.1. DVL1 and 3 Are Highly Expressed in Retinal Capillaries, Co-Localizing with the Tight Junction Proteins, CLDN5 and ZO-1
3.2. Dvl1 Knockdown Reduced Basal Barrier Properties despite Increased β-Catenin Signaling
3.3. Knockdown of Dvl1 but Not Dvl3, Ablated Norrin-Induced Blood–Retinal Barrier Restoration after VEGF
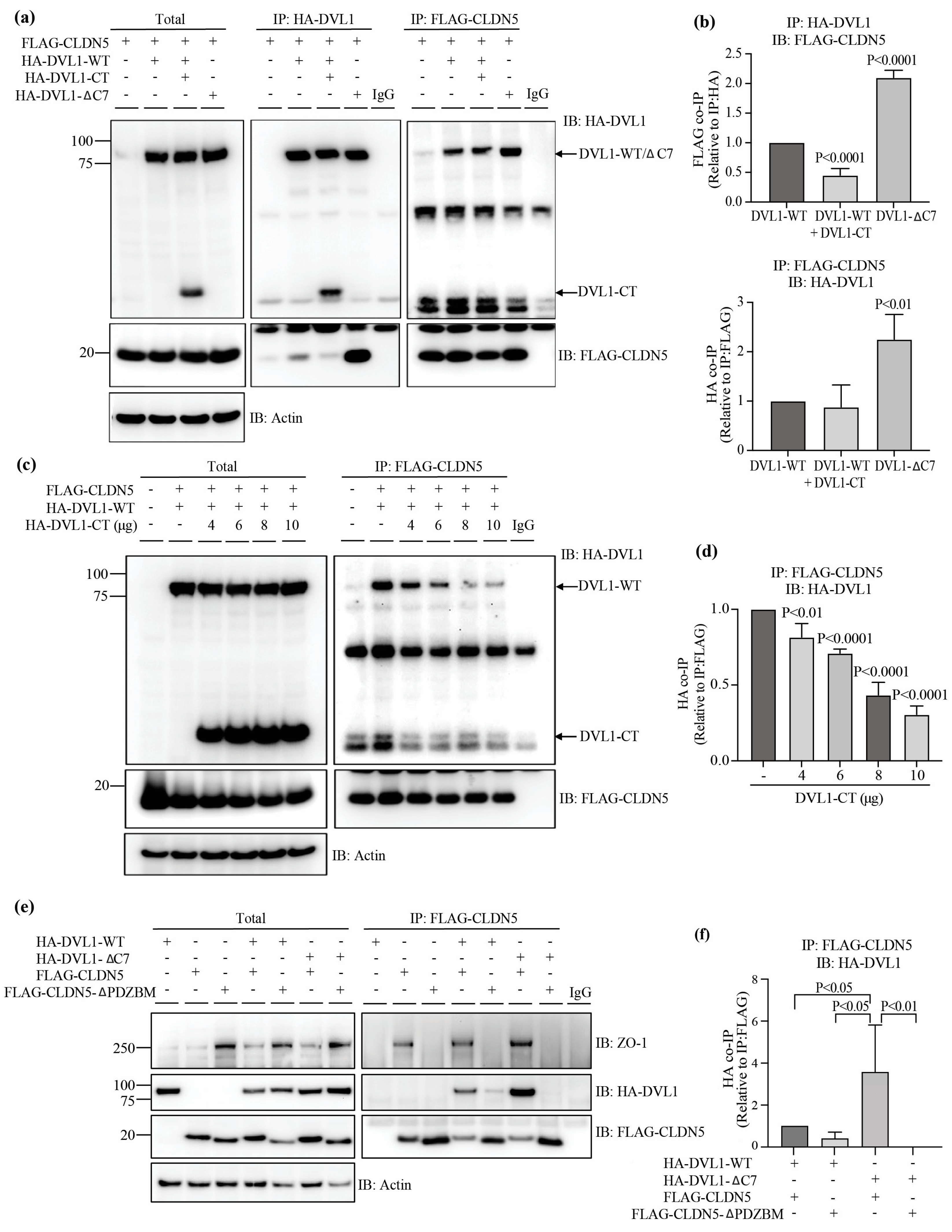
3.4. DVL1 Interacts with CLDN5 and ZO-1, While DVL3 Interacts with ZO-1
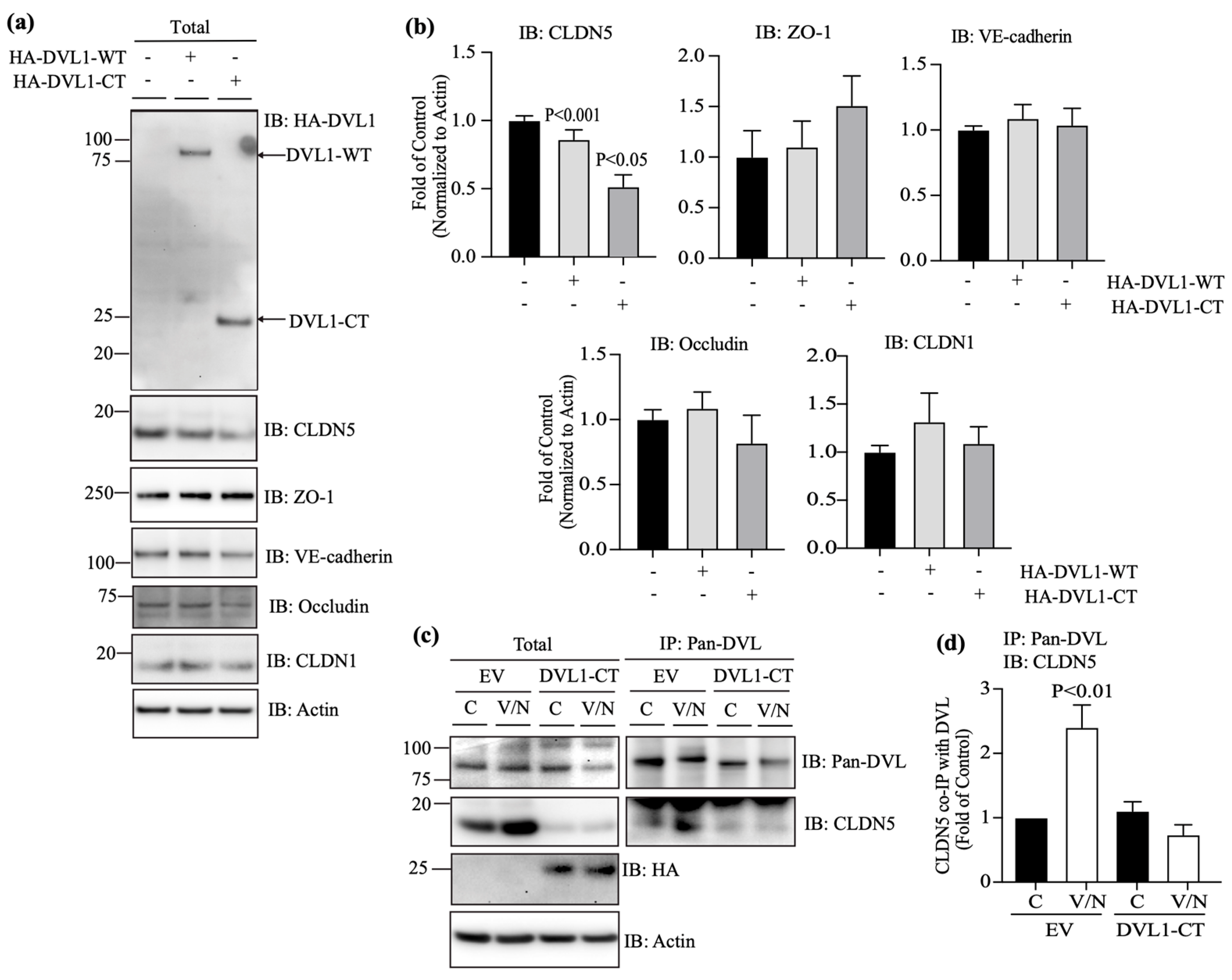
3.5. DVL1 and CLDN5 Interaction Is Required for Basal and Norrin-Induced Endothelial Barrier Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Wang, Y.; Tischfield, M.; Williams, J.; Smallwood, P.M.; Rattner, A.; Taketo, M.M.; Nathans, J. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Investig. 2014, 124, 3825–3846. [Google Scholar] [CrossRef]
- Scha¨fer, N.F.; Luhmann, U.F.O.; Feil, S.; Berger, W. Differential gene expression in Ndph-knockout mice in retinal development. Investig. Opthalmology Vis. Sci. 2009, 50, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, J.; Fritzsche, M.; Feil, S.; Mohn, L.; Berger, W. Norrin stimulates cell proliferation in the superficial retinal vascular plexus and is pivotal for the recruitment of mural cells. Hum. Mol. Genet. 2012, 21, 2619–2630. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.C.; Feng, Y.; Sothilingam, V.; Garrido, M.G.; Tanimoto, N.; Acar, N.; Shan, S.; Seebauer, B.; Berger, W.; Hammes, H.-P.; et al. Long-term consequences of developmental vascular defects on retinal vessel homeostasis and function in a mouse model of Norrie disease. PLoS ONE 2017, 12, e0178753. [Google Scholar] [CrossRef]
- Ngo, M.H.; Borowska-Fielding, J.; Heathcote, G.; Nejat, S.; Kelly, M.E.; McMaster, C.R.; Robitaille, J.M. Fzd4 Haploinsufficiency Delays Retinal Revascularization in the Mouse Model of Oxygen Induced Retinopathy. PLoS ONE 2016, 11, e0158320. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular development in the retina and inner ear: Control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 2004, 116, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Paes, K.T.; Wang, E.; Henze, K.; Vogel, P.; Read, R.; Suwanichkul, A.; Kirkpatrick, L.L.; Potter, D.; Newhouse, M.M.; Rice, D.S. Frizzled 4 is required for retinal angiogenesis and maintenance of the blood-retina barrier. Investig. Opthalmol. Vis. Sci. 2011, 52, 6452–6461. [Google Scholar] [CrossRef]
- Xia, C.-H.; Liu, H.; Cheung, D.; Wang, M.; Cheng, C.; Du, X.; Chang, B.; Beutler, B.; Gong, X. A model for familial exudative vitreoretinopathy caused by LPR5 mutations. Hum. Mol. Genet. 2008, 17, 1605–1612. [Google Scholar] [CrossRef]
- Chen, J.; Stahl, A.; Krah, N.M.; Seaward, M.R.; Dennison, R.J.; Sapieha, P.; Hua, J.; Hatton, C.J.; Juan, A.M.; Aderman, C.M.; et al. Wnt signaling mediates pathological vascular growth in proliferative retinopathy. Circulation 2011, 124, 1871–1881. [Google Scholar] [CrossRef]
- Junge, H.J.; Yang, S.; Burton, J.B.; Paes, K.; Shu, X.; French, D.M.; Costa, M.; Rice, D.S.; Ye, W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009, 139, 299–311. [Google Scholar] [CrossRef]
- Zhang, C.; Lai, M.B.; Pedler, M.G.; Johnson, V.; Adams, R.H.; Petrash, J.M.; Chen, Z.; Junge, H.J. Endothelial Cell-Specific Inactivation of TSPAN12 (Tetraspanin 12) Reveals Pathological Consequences of Barrier Defects in an Otherwise Intact Vasculature. Arter. Thromb. Vasc. Biol. 2018, 38, 2691–2705. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Wang, Y.; Cahill, H.; Yu, M.; Badea, T.C.; Smallwood, P.M.; Peachey, N.S.; Nathans, J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell 2009, 139, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Smallwood, P.; Nathans, J. Expression of the Norrie disease gene (Ndp) in developing and adult mouse eye, ear, and brain. Gene Expr. Patterns 2011, 11, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; van de Pol, D.; Bächner, D.; Oerlemans, F.; Winkens, H.; Hameister, H.; Wieringa, B.; Hendriks, W.; Ropers, H.-H. An animal model for Norrie disease (ND): Gene targeting of the mouse ND gene. Hum. Mol. Genet. 1996, 5, 51–59. [Google Scholar] [CrossRef]
- Lee, H.; Jo, D.H.; Kim, J.H.; Kim, J.H. Norrin expression in endothelial cells in the developing mouse retina. Acta Histochem. 2013, 115, 447–451. [Google Scholar] [CrossRef]
- Ke, J.; Harikumar, K.G.; Erice, C.; Chen, C.; Gu, X.; Wang, L.; Parker, N.; Cheng, Z.; Xu, W.; Williams, B.O.; et al. Structure and function of Norrin in assembly and activation of a Frizzled 4-Lrp5/6 complex. Genes Dev. 2013, 27, 2305–2319. [Google Scholar] [CrossRef]
- DeBruine, Z.J.; Xu, H.E.; Melcher, K. Assembly and architecture of the Wnt/beta-catenin signalosome at the membrane. Br. J. Pharmacol. 2017, 174, 4564–4574. [Google Scholar] [CrossRef]
- Gammons, M.V.; Renko, M.; Johnson, C.M.; Rutherford, T.J.; Bienz, M. Wnt Signalosome Assembly by DEP Domain Swapping of Dishevelled. Mol. Cell 2016, 64, 92–104. [Google Scholar] [CrossRef]
- Mahoney, J.P.; Bruguera, E.S.; Vasishtha, M.; Killingsworth, L.B.; Kyaw, S.; Weis, W.I. PI(4,5)P(2)-stimulated positive feedback drives the recruitment of Dishevelled to Frizzled in Wnt-beta-catenin signaling. Sci. Signal. 2022, 15, eabo2820. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Cattelino, A.; Liebner, S.; Gallini, R.; Zanetti, A.; Balconi, G.; Corsi, A.; Bianco, P.; Wolburg, H.; Moore, R.; Oreda, B.; et al. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J. Cell Biol. 2003, 162, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chen, Y.-G. Dishevelled: The hub of Wnt signaling. Cell. Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef]
- Lijam, N.; Paylor, R.; McDonald, M.P.; Crawley, J.N.; Deng, C.X.; Herrup, K.; Stevens, K.E.; Maccaferri, G.; McBain, C.J.; Sussman, D.J.; et al. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell 1997, 90, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; LaPorte, P.; Paylor, R.; Wynshaw-Boris, A. Expanded characterization of the social interaction abnormalities in mice lacking Dvl1. Genes Brain Behav. 2004, 3, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Etheridge, S.L.; Ray, S.; Li, S.; Hamblet, N.S.; Lijam, N.; Tsang, M.; Greer, J.; Kardos, N.; Wang, J.; Sussman, D.J.; et al. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008, 4, e1000259. [Google Scholar] [CrossRef]
- Lee, H.J.; Shi, D.L.; Zheng, J.J. Conformational change of Dishevelled plays a key regulatory role in the Wnt signaling pathways. eLife 2015, 4, e08142. [Google Scholar] [CrossRef]
- Diaz-Coranguez, M.; Lin, C.M.; Liebner, S.; Antonetti, D.A. Norrin restores blood-retinal barrier properties after vascular endothelial growth factor-induced permeability. J. Biol. Chem. 2020, 295, 4647–4660. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Wolpert, E.B. Isolation and characterization of retinal endothelial cells. Methods Mol. Med. 2003, 89, 365–374. [Google Scholar]
- Tiruppathi, C.; Malik, A.B.; Del Vecchio, P.J.; Keese, C.R.; Giaever, I. Electrical method for detection of endothelial cell shape change in real time: Assessment of endothelial barrier function. Proc. Natl. Acad. Sci. USA 1992, 89, 7919–7923. [Google Scholar] [CrossRef]
- Harhaj, N.S.; Barber, A.J.; Antonetti, D.A. Platelet-derived growth factor mediates si redistribution and increases permeability in MDCK cells. J. Cell Physiol. 2002, 193, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrosio, A.F.; Antonetti, D.A. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010, 59, 2872–2882. [Google Scholar] [CrossRef] [PubMed]
- Bernatík, O.; Šedová, K.; Schille, C.; Ganji, R.S.; Červenka, I.; Trantírek, L.; Schambony, A.; Zdráhal, Z.; Bryja, V. Functional analysis of dishevelled-3 phosphorylation identifies distinct mechanisms driven by casein kinase 1ϵ and frizzled5. J. Biol. Chem. 2014, 289, 23520–23533. [Google Scholar] [CrossRef] [PubMed]
- Hanáková, K.; Bernatík, O.; Kravec, M.; Micka, M.; Kumar, J.; Harnoš, J.; Ovesná, P.; Paclíková, P.; Rádsetoulal, M.; Potěšil, D.; et al. Comparative phosphorylation map of Dishevelled 3 links phospho-signatures to biological outputs. Cell Commun. Signal. 2019, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Lee, H.J.; Saquet, A.; Cheng, X.N.; Shao, M.; Zheng, J.J.; Shi, D.L. Autoinhibition of Dishevelled protein regulated by its extreme C terminus plays a distinct role in Wnt/beta-catenin and Wnt/planar cell polarity (PCP) signaling pathways. J. Biol. Chem. 2017, 292, 5898–5908. [Google Scholar] [CrossRef] [PubMed]
- Harnoš, J.; Cañizal, M.C.A.; Jurásek, M.; Kumar, J.; Holler, C.; Schambony, A.; Hanáková, K.; Bernatík, O.; Zdráhal, Z.; Gömöryová, K.; et al. Dishevelled-3 conformation dynamics analyzed by FRET-based biosensors reveals a key role of casein kinase 1. Nat. Commun. 2019, 10, 1804. [Google Scholar] [CrossRef]
- Li, L.; Mao, B.; Yan, M.; Wu, S.; Ge, R.; Lian, Q.; Cheng, C.Y. Planar cell polarity protein Dishevelled 3 (Dvl3) regulates ectoplasmic specialization (ES) dynamics in the testis through changes in cytoskeletal organization. Cell Death Dis. 2019, 10, 194. [Google Scholar] [CrossRef]
- Wang, Y.; Sabbagh, M.F.; Gu, X.; Rattner, A.; Williams, J.; Nathans, J. Beta-catenin signaling regulates barrier-specific gene expression in circumventricular organ and ocular vasculatures. eLife 2019, 8, e43257. [Google Scholar] [CrossRef]
- Devenport, D. The cell biology of planar cell polarity. J. Cell Biol. 2014, 207, 171–179. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, J.; Yang, G.-Y.; Wang, Q.-J.; Qian, L.; Chen, Y.-M.; Chen, F.; Tao, Y.; Hu, H.-S.; Wang, T.; et al. Dishevelled promotes axon differentiation by regulating atypical protein kinase C. Nat. Cell Biol. 2007, 9, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Cirone, P.; Lin, S.; Griesbach, H.L.; Zhang, Y.; Slusarski, D.C.; Crews, C.M. A role for planar cell polarity signaling in angiogenesis. Angiogenesis 2008, 11, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Sewduth, R.N.; Kovacic, H.; Jaspard-Vinassa, B.; Jecko, V.; Wavasseur, T.; Fritsch, N.; Pernot, M.; Jeaningros, S.; Roux, E.; Dufourcq, P.; et al. PDZRN3 destabilizes endothelial cell-cell junctions through a PKCzeta-containing polarity complex to increase vascular permeability. Sci. Signal. 2017, 10, eaag3209. [Google Scholar] [CrossRef] [PubMed]
- Sewduth, R.N.; Jaspard-Vinassa, B.; Peghaire, C.; Guillabert, A.; Franzl, N.; Larrieu-Lahargue, F.; Moreau, C.; Fruttiger, M.; Dufourcq, P.; Couffinhal, T.; et al. The ubiquitin ligase PDZRN3 is required for vascular morphogenesis through Wnt/planar cell polarity signalling. Nat. Commun. 2014, 5, 4832. [Google Scholar] [CrossRef]
- Fanning, A.S.; Anderson, J.M. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Coránguez, M.; González-González, L.; Wang, A.; Liu, X.; Antonetti, D.A. Disheveled-1 Interacts with Claudin-5 and Contributes to Norrin-Induced Endothelial Barrier Restoration. Cells 2023, 12, 2402. https://doi.org/10.3390/cells12192402
Díaz-Coránguez M, González-González L, Wang A, Liu X, Antonetti DA. Disheveled-1 Interacts with Claudin-5 and Contributes to Norrin-Induced Endothelial Barrier Restoration. Cells. 2023; 12(19):2402. https://doi.org/10.3390/cells12192402
Chicago/Turabian StyleDíaz-Coránguez, Mónica, Laura González-González, Amy Wang, Xuwen Liu, and David A. Antonetti. 2023. "Disheveled-1 Interacts with Claudin-5 and Contributes to Norrin-Induced Endothelial Barrier Restoration" Cells 12, no. 19: 2402. https://doi.org/10.3390/cells12192402
APA StyleDíaz-Coránguez, M., González-González, L., Wang, A., Liu, X., & Antonetti, D. A. (2023). Disheveled-1 Interacts with Claudin-5 and Contributes to Norrin-Induced Endothelial Barrier Restoration. Cells, 12(19), 2402. https://doi.org/10.3390/cells12192402