

Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit?

 ,
, 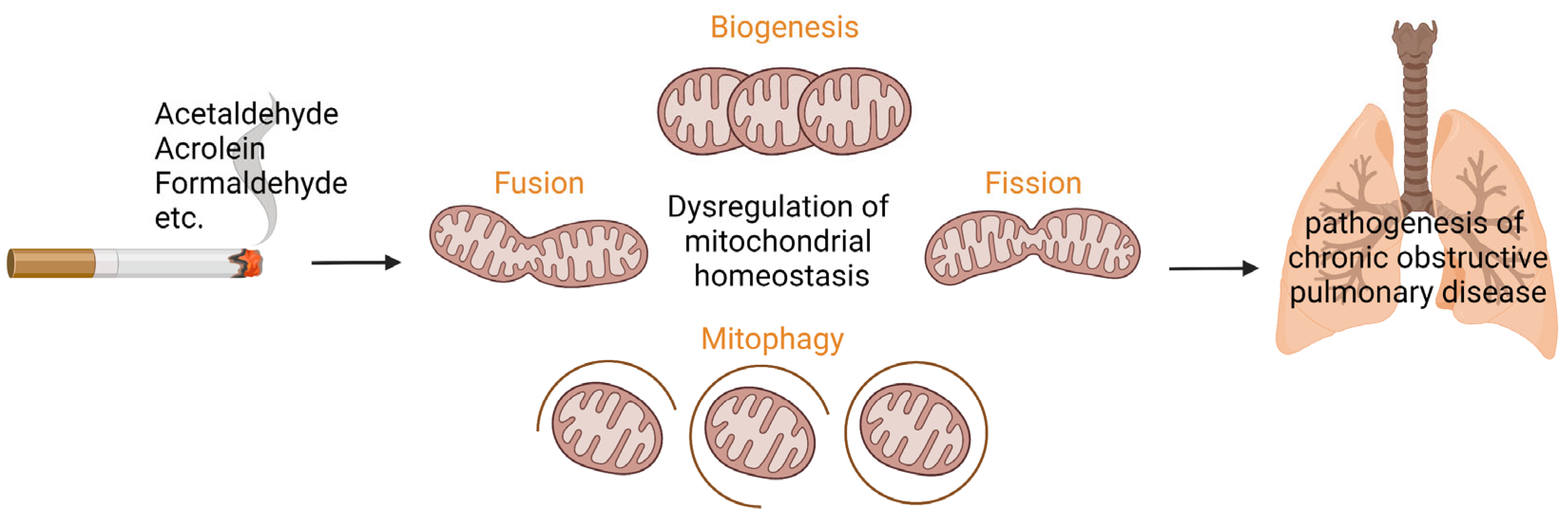{kind=link}
Abstract
:1. Aldehydes as a Component of Cigarette Smoke
2. Aldehyde-Induced Respiratory Toxicity Is Associated with the Development of Chronic Obstructive Pulmonary Disease
3. Mitochondrial Function in Healthy Cells of the Airways and Lungs
4. Mitochondrial Abnormalities in Epithelial Cells of the Airways and Lungs from COPD Patients
5. CS-Induced Mitochondrial Dysfunction in Epithelial Cells of the Airways and Lungs
6. Aldehydes-Induced Mitochondrial Dysfunction in Lung Cells Associated with COPD Pathogenesis
7. Potential Therapeutical Applications Targeting Aldehyde-Induced Mitochondrial Dysfunction in COPD
7.1. Mitochondria as Therapeutic Target in COPD
7.2. Targeting Aldehydes
8. Regulation of Aldehydes in CS
8.1. Implications for Regulation
8.2. Implications for Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Factsheet Tobacco. Available online: https://www.who.int/news-room/fact-sheets/detail/tobacco (accessed on 18 July 2022).
- Burns, D.M.; Dybing, E.; Gray, N.; Hecht, S.; Anderson, C.; Sanner, T.; O’Connor, R.; Djordjevic, M.; Dresler, C.; Hainaut, P.; et al. Mandated lowering of toxicants in cigarette smoke: A description of the World Health Organization TobReg proposal. Tob. Control. 2008, 17, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization; World Health Organization Tobacco Free Initiative. The Scientific Basis of Tobacco Product Regulation: Second Report of a WHO Study Group; WHO: Geneva, Switzerland, 2008. [Google Scholar]
- Talhout, R.; Schulz, T.; Florek, E.; van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Rodgman, A.; Perfetti, T.A. The Chemical Components of Tobacco and Tobacco Smoke; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Cheah, N.P. Volatile aldehydes in tobacco smoke: Source fate and risk. Ph.D. Thesis, Maastricht University, Maastricht, The Netherlands, 2016. [Google Scholar]
- LoPachin, R.M.; Gavin, T. Molecular mechanisms of aldehyde toxicity: A chemical perspective. Chem. Res. Toxicol. 2014, 27, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Corley, R.A.; Kabilan, S.; Kuprat, A.P.; Carson, J.P.; Jacob, R.E.; Minard, K.R.; Teeguarden, J.G.; Timchalk, C.; Pipavath, S.; Glenny, R.; et al. Comparative Risks of Aldehyde Constituents in Cigarette Smoke Using Transient Computational Fluid Dynamics/Physiologically Based Pharmacokinetic Models of the Rat and Human Respiratory Tracts. Toxicol. Sci. 2015, 146, 65–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowles, J.; Dybing, E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob. Control. 2003, 12, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhout, R.; Opperhuizen, A.; van Amsterdam, J.G. Sugars as tobacco ingredient: Effects on mainstream smoke composition. Food. Chem. Toxicol. 2006, 44, 1789–1798. [Google Scholar] [CrossRef]
- Fagerson, I.S. Thermal degradation of carbohydrates; a review. J. Agric. Food Chem. 1969, 17, 747–750. [Google Scholar] [CrossRef]
- Mattonai, M.; Tamburini, D.; Colombini, M.P.; Ribechini, E. Timing in Analytical Pyrolysis: Py(HMDS)-GC/MS of Glucose and Cellulose Using Online Micro Reaction Sampler. Anal. Chem. 2016, 88, 9318–9325. [Google Scholar] [CrossRef]
- Mattonai, M.; Ribechini, E. A comparison of fast and reactive pyrolysis with insitu derivatisation of fructose, inulin and Jerusalem artichoke (Helianthus tuberosus). Anal. Chim. Acta 2018, 1017, 66–74. [Google Scholar] [CrossRef]
- Roemer, E.; Schorp, M.K.; Piadé, J.J.; Seeman, J.I.; Leyden, D.E.; Haussmann, H.J. Scientific assessment of the use of sugars as cigarette tobacco ingredients: A review of published and other publicly available studies. Crit. Rev. Toxicol. 2012, 42, 244–278. [Google Scholar] [CrossRef]
- Leffingwell, J. BA Basic Chemical Constituents of Tobacco Leaf and Differences among Tobacco Types; Blackwell Science: Oxford, UK, 1999; pp. 265–284. [Google Scholar]
- Cahours, X.; Verron, T.; Purkis, S. Effect of sugar content on acetaldehyde yield in cigarette smoke. Beiträge Zur Tab. Int./Contrib. Tob. Res. 2012, 25, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Seeman, J.I.; Laffoon, S.W.; Kassman, A.J. Evaluation of relationships between mainstream smoke acetaldehyde and "tar" and carbon monoxide yields in tobacco smoke and reducing sugars in tobacco blends of U.S. commercial cigarettes. Inhal. Toxicol. 2003, 15, 373–395. [Google Scholar] [CrossRef]
- Pauwels, C.; Klerx, W.N.M.; Pennings, J.L.A.; Boots, A.W.; van Schooten, F.J.; Opperhuizen, A.; Talhout, R. Cigarette Filter Ventilation and Smoking Protocol Influence Aldehyde Smoke Yields. Chem. Res. Toxicol. 2018, 31, 462–471. [Google Scholar] [CrossRef]
- Sleiman, M.; Logue, J.M.; Montesinos, V.N.; Russell, M.L.; Litter, M.I.; Gundel, L.A.; Destaillats, H. Emissions from Electronic Cigarettes: Key Parameters Affecting the Release of Harmful Chemicals. Environ. Sci. Technol. 2016, 50, 9644–9651. [Google Scholar] [CrossRef] [Green Version]
- Sinharoy, P.; McAllister, S.L.; Vasu, M.; Gross, E.R. Environmental Aldehyde Sources and the Health Implications of Exposure. Adv. Exp. Med. Biol. 2019, 1193, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Ogunwale, M.A.; Li, M.; Ramakrishnam Raju, M.V.; Chen, Y.; Nantz, M.H.; Conklin, D.J.; Fu, X.-A. Aldehyde detection in electronic cigarette aerosols. ACS Omega 2017, 2, 1207–1214. [Google Scholar] [CrossRef]
- Fagan, P.; Pokhrel, P.; Herzog, T.A.; Moolchan, E.T.; Cassel, K.D.; Franke, A.A.; Li, X.; Pagano, I.; Trinidad, D.R.; Sakuma, K.K.; et al. Sugar and Aldehyde Content in Flavored Electronic Cigarette Liquids. Nicotine Tob. Res. 2018, 20, 985–992. [Google Scholar] [CrossRef]
- Ruszkiewicz, J.A.; Zhang, Z.; Gonçalves, F.M.; Tizabi, Y.; Zelikoff, J.T.; Aschner, M. Neurotoxicity of e-cigarettes. Food Chem. Toxicol. 2020, 138, 111245. [Google Scholar] [CrossRef]
- Kosmider, L.; Sobczak, A.; Fik, M.; Knysak, J.; Zaciera, M.; Kurek, J.; Goniewicz, M.L. Carbonyl compounds in electronic cigarette vapors: Effects of nicotine solvent and battery output voltage. Nicotine Tob. Res. 2014, 16, 1319–1326. [Google Scholar] [CrossRef]
- Goniewicz, M.L.; Knysak, J.; Gawron, M.; Kosmider, L.; Sobczak, A.; Kurek, J.; Prokopowicz, A.; Jablonska-Czapla, M.; Rosik-Dulewska, C.; Havel, C.; et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob. Control. 2014, 23, 133–139. [Google Scholar] [CrossRef]
- Farsalinos, K.E.; Voudris, V.; Poulas, K. E-cigarettes generate high levels of aldehydes only in ‘dry puff’ conditions. Addiction 2015, 110, 1352–1356. [Google Scholar] [CrossRef] [PubMed]
- Faroon, O.; Roney, N.; Taylor, J.; Ashizawa, A.; Lumpkin, M.H.; Plewak, D.J. Acrolein environmental levels and potential for human exposure. Toxicol. Ind. Health 2008, 24, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Rietjens, I.; Michael, A.; Bolt, H.M.; Siméon, B.; Andrea, H.; Nils, H.; Christine, K.; Angela, M.; Gloria, P.; Daniel, R.; et al. The role of endogenous versus exogenous sources in the exposome of putative genotoxins and consequences for risk assessment. Arch. Toxicol. 2022, 96, 1297–1352. [Google Scholar] [CrossRef] [PubMed]
- Kuykendall, J.R. 8.16-Aldehydes. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier: Oxford, UK, 2010; pp. 291–330. [Google Scholar]
- Xie, Z.; Baba, S.P.; Sweeney, B.R.; Barski, O.A. Detoxification of aldehydes by histidine-containing dipeptides: From chemistry to clinical implications. Chem. Biol. Interact. 2013, 202, 288–297. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, P.J.; Siraki, A.G.; Shangari, N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005, 35, 609–662. [Google Scholar] [CrossRef]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, J.; Holley, D.W.; Kawamoto, T.; Bultman, S.J. The failure of two major formaldehyde catabolism enzymes (ADH5 and ALDH2) leads to partial synthetic lethality in C57BL/6 mice. Genes Environ. 2020, 42, 21. [Google Scholar] [CrossRef]
- Ma, I.; Allan, A.L. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. Rep. 2011, 7, 292–306. [Google Scholar] [CrossRef]
- Hegab, A.E.; Ha, V.L.; Bisht, B.; Darmawan, D.O.; Ooi, A.T.; Zhang, K.X.; Paul, M.K.; Kim, Y.S.; Gilbert, J.L.; Attiga, Y.S.; et al. Aldehyde dehydrogenase activity enriches for proximal airway basal stem cells and promotes their proliferation. Stem Cells Dev. 2014, 23, 664–675. [Google Scholar] [CrossRef]
- Hegab, A.E.; Ha, V.L.; Darmawan, D.O.; Gilbert, J.L.; Ooi, A.T.; Attiga, Y.S.; Bisht, B.; Nickerson, D.W.; Gomperts, B.N. Isolation and in vitro characterization of basal and submucosal gland duct stem/progenitor cells from human proximal airways. Stem Cells Transl. Med. 2012, 1, 719–724. [Google Scholar] [CrossRef]
- Dipple, K.M.; Crabb, D.W. The mitochondrial aldehyde dehydrogenase gene resides in an HTF island but is expressed in a tissue-specific manner. Biochem. Biophys. Res. Commun. 1993, 193, 420–427. [Google Scholar] [CrossRef]
- Costa, D.L.; Kutzman, R.S.; Lehmann, J.R.; Drew, R.T. Altered lung function and structure in the rat after subchronic exposure to acrolein. Am. Rev. Respir. Dis. 1986, 133, 286–291. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef] [Green Version]
- Zarkovic, N.; Cipak, A.; Jaganjac, M.; Borovic, S.; Zarkovic, K. Pathophysiological relevance of aldehydic protein modifications. J. Proteomics. 2013, 92, 239–247. [Google Scholar] [CrossRef]
- IARC (International Agency for Research on Cancer). Acrolein, Crotonaldehyde, and Arecoline; IARC Working Group: Lyon, France, 2020. [Google Scholar]
- European Commission. Commission Directive (EU) 2017/164 of 31 January 2017 establishing a fourth list of indicative occupational exposure limit values pursuant to Council Directive 98/24/EC, and amending Commission Directives 91/322/EEC, 2000/39/EC and 2009/161/EU. Off. J. Eur. Union 2017, L 27, 115–120. [Google Scholar]
- ACGIH (American Conference of Governmental Industrial Hygienists). Threshold Limit Values for Chemical Substances and Physical Agents and Biological Exposure Indices; ACGIH: Cincinnati, OH, USA, 2019. [Google Scholar]
- Yeager, R.P.; Kushman, M.; Chemerynski, S.; Weil, R.; Fu, X.; White, M.; Callahan-Lyon, P.; Rosenfeldt, H. Proposed Mode of Action for Acrolein Respiratory Toxicity Associated with Inhaled Tobacco Smoke. Toxicol. Sci. 2016, 151, 347–364. [Google Scholar] [CrossRef]
- Moretto, N.; Volpi, G.; Pastore, F.; Facchinetti, F. Acrolein effects in pulmonary cells: Relevance to chronic obstructive pulmonary disease. Ann. N. Y. Acad. Sci. 2012, 1259, 39–46. [Google Scholar] [CrossRef]
- Deshmukh, H.S.; Shaver, C.; Case, L.M.; Dietsch, M.; Wesselkamper, S.C.; Hardie, W.D.; Korfhagen, T.R.; Corradi, M.; Nadel, J.A.; Borchers, M.T.; et al. Acrolein-activated matrix metalloproteinase 9 contributes to persistent mucin production. Am. J. Respir. Cell Mol. Biol. 2008, 38, 446–454. [Google Scholar] [CrossRef] [Green Version]
- IARC (International Agency for Research on Cancer). Chemical Agents and Related Occupations; IARC Working Group: Lyon, France, 2012. [Google Scholar]
- IARC (International Agency for Research on Cancer). Allyl Compounds, Aldehydes, Epoxides and Peroxides; IARC Working Group: Lyon, France, 1985. [Google Scholar]
- IARC (International Agency for Research on Cancer). Overall evaluations of carcinogenicity: An updating of IARC Monographs volumes 1 to 42. IARC Monogr. Eval. Carcinog. Risk Chem. Hum. Suppl. 1987, 7, 1–440. [Google Scholar]
- ECHA (European Chemicals Agency). Worker Exposure to Formaldehyde and Formaldehyde Releasers; European Chemicals Agency: Helsinki, Finland, 2019; p. 59. [Google Scholar]
- OSHA (Occupational Safety and Health Administration). OSHA Occupational Chemical Database Acetaldehyde; Occupational Safety and Health Administration: Washington, DC, USA, 2020. [Google Scholar]
- Cheah, N.P.; Pennings, J.L.; Vermeulen, J.P.; van Schooten, F.J.; Opperhuizen, A. In vitro effects of aldehydes present in tobacco smoke on gene expression in human lung alveolar epithelial cells. Toxicol. Vitr. 2013, 27, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, L.; Barbosa, E.; Charão, M.F.; Brucker, N. Formaldehyde toxicity reports from in vitro and in vivo studies: A review and updated data. Drug Chem. Toxicol. 2022, 45, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Wilk, J.B.; Shrine, N.R.; Loehr, L.R.; Zhao, J.H.; Manichaikul, A.; Lopez, L.M.; Smith, A.V.; Heckbert, S.R.; Smolonska, J.; Tang, W.; et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am. J. Respir. Crit. Care Med. 2012, 186, 622–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, D.B.; Eijgelsheim, M.; Wilk, J.B.; Gharib, S.A.; Loehr, L.R.; Marciante, K.D.; Franceschini, N.; van Durme, Y.M.; Chen, T.H.; Barr, R.G.; et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat. Genet. 2010, 42, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repapi, E.; Sayers, I.; Wain, L.V.; Burton, P.R.; Johnson, T.; Obeidat, M.; Zhao, J.H.; Ramasamy, A.; Zhai, G.; Vitart, V.; et al. Genome-wide association study identifies five loci associated with lung function. Nat. Genet. 2010, 42, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Xia, P.; Yan, L.; Gou, X.; Giesy, J.P.; Dai, J.; Yu, H.; Zhang, X. Toxicological Mechanism of Individual Susceptibility to Formaldehyde-Induced Respiratory Effects. Environ. Sci. Technol. 2022, 56, 6511–6524. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. Acetaldehyde: Hazard. Summary-Created in April 1992; Revised in January 2000; U.S. Environmental Protection Agency: Washington, DC, USA, 2000. [Google Scholar]
- U.S. Environmental Protection Agency. Health Assessment Document for Acetaldehyde; Environmental Criteria and Assessment Office, Office of Health and Environmental Assessment, Office of Research and Development: Research Triangle Park, NC, USA, 1987. [Google Scholar]
- U.S. Department of Health and Human Services. Registry of Toxic Effects of Chemical Substances (RTECS, Online Database); National Toxicology Information Program, National Library of Medicine: Bethesda, MD, USA, 1993. [Google Scholar]
- U.S. Environmental Protection Agency. Integrated Risk Information System (IRIS) on Acetaldehyde; National Center for Environmental Assessment, Office of Research and Development: Washington, DC, USA, 1999. [Google Scholar]
- Feron, V.; Kruysse, A.; Woutersen, R. Respiratory tract tumours in hamsters exposed to acetaldehyde vapour alone or simultaneously to benzo (a) pyrene or diethylnitrosamine. Eur. J. Cancer Clin. Oncol. 1982, 18, 13–31. [Google Scholar] [CrossRef]
- Kruysse, A.; Feron, V.J.; Til, H.P. Repeated exposure to acetaldehyde vapor: Studies in Syrian golden hamsters. Arch. Environ. Health An. Int. J. 1975, 30, 449–452. [Google Scholar] [CrossRef]
- Feron, V. Effects of Exposure to Acetaldehyde in Syrian Hamsters Simultaneously Treated with Benzo (a) pyrene or Diethylnitrosamine1. In The Syrian Hamster in Toxicology and Carcinogenesis Research; Karger Publishers: London, UK, 1979; Volume 24, pp. 162–176. [Google Scholar]
- National Research Council (US) Committee. Emergency and Continuous Exposure Guidance Levels for Selected Submarine Contaminants; National Academies Press (US): Washington, DC, USA, 2009; Volume 3. [Google Scholar]
- IARC (International Agency for Research on Cancer). Acetaldehyde. Re-Evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part. Two); IARC: Lyon, France, 1999; pp. 319–336. [Google Scholar]
- Van der Toorn, M.; Slebos, D.J.; de Bruin, H.G.; Gras, R.; Rezayat, D.; Jorge, L.; Sandra, K.; van Oosterhout, A.J. Critical role of aldehydes in cigarette smoke-induced acute airway inflammation. Respir. Res. 2013, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, H.; Wang, A.; Liu, Y.; Hou, H.; Hu, Q. Combined effects of co-exposure to formaldehyde and acrolein mixtures on cytotoxicity and genotoxicity in vitro. Environ. Sci. Pollut. Res. Int. 2018, 25, 25306–25314. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, J.; Chen, H.; Wang, A.; Liu, Y.; Hou, H.; Hu, Q. Combined cytotoxicity of co-exposure to aldehyde mixtures on human bronchial epithelial BEAS-2B cells. Environ. Pollut. 2019, 250, 650–661. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, J.; Cheng, W.; Chen, H.; Wang, A.; Liu, Y.; Hou, H.; Hu, Q. Combined cell death of co-exposure to aldehyde mixtures on human bronchial epithelial BEAS-2B cells: Molecular insights into the joint action. Chemosphere 2020, 244, 125482. [Google Scholar] [CrossRef]
- Zulueta, A.; Caretti, A.; Campisi, G.M.; Brizzolari, A.; Abad, J.L.; Paroni, R.; Signorelli, P.; Ghidoni, R. Inhibitors of ceramide de novo biosynthesis rescue damages induced by cigarette smoke in airways epithelia. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 753–759. [Google Scholar] [CrossRef]
- Corradi, M.; Pignatti, P.; Manini, P.; Andreoli, R.; Goldoni, M.; Poppa, M.; Moscato, G.; Balbi, B.; Mutti, A. Comparison between exhaled and sputum oxidative stress biomarkers in chronic airway inflammation. Eur. Respir. J. 2004, 24, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Yasuo, M.; Droma, Y.; Kitaguchi, Y.; Ito, M.; Imamura, H.; Kawakubo, M.; Hanaoka, M. The relationship between acrolein and oxidative stress in COPD: In systemic plasma and in local lung tissue. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1527–1537. [Google Scholar] [CrossRef] [Green Version]
- Tu, C.; Mammen, M.J.; Li, J.; Shen, X.; Jiang, X.; Hu, Q.; Wang, J.; Sethi, S.; Qu, J. Large-scale, ion-current-based proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease patients. J. Proteome Res. 2014, 13, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Masuda, N.; Oniki, K.; Saruwatari, J.; Kajiwara, A.; Otake, K.; Ogata, Y.; Nakagawa, K. Association between the aldehyde dehydrogenase 2*2 allele and smoking-related chronic airway obstruction in a Japanese general population: A pilot study. Toxicol. Lett. 2015, 236, 117–122. [Google Scholar] [CrossRef]
- Kuroda, A.; Hegab, A.E.; Jingtao, G.; Yamashita, S.; Hizawa, N.; Sakamoto, T.; Yamada, H.; Suzuki, S.; Ishii, M.; Namkoong, H.; et al. Effects of the common polymorphism in the human aldehyde dehydrogenase 2 (ALDH2) gene on the lung. Respir. Res. 2017, 18, 69. [Google Scholar] [CrossRef] [Green Version]
- Oyama, T.; Isse, T.; Ogawa, M.; Muto, M.; Uchiyama, I.; Kawamoto, T. Susceptibility to inhalation toxicity of acetaldehyde in Aldh2 knockout mice. Front. Biosci.-Landmark 2007, 12, 1927–1934. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Bruse, S.; Liu, Y.; Duffy, V.; Zhang, C.; Oyamada, N.; Randell, S.; Matsumoto, A.; Thompson, D.C.; Lin, Y.; et al. Aldehyde dehydrogenase 3A1 protects airway epithelial cells from cigarette smoke-induced DNA damage and cytotoxicity. Free Radic. Biol. Med. 2014, 68, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloonan, S.M.; Kim, K.; Esteves, P.; Trian, T.; Barnes, P.J. Mitochondrial dysfunction in lung ageing and disease. Eur. Respir. Rev. 2020, 29, 200165. [Google Scholar] [CrossRef] [PubMed]
- Aghapour, M.; Remels, A.H.V.; Pouwels, S.D.; Bruder, D.; Hiemstra, P.S.; Cloonan, S.M.; Heijink, I.H. Mitochondria: At the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L149–L164. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Gomes, L.C.; Scorrano, L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta 2013, 1833, 205–212. [Google Scholar] [CrossRef]
- Fritsch, L.E.; Moore, M.E.; Sarraf, S.A.; Pickrell, A.M. Ubiquitin and Receptor-Dependent Mitophagy Pathways and Their Implication in Neurodegeneration. J. Mol. Biol. 2020, 432, 2510–2524. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [Green Version]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef]
- Haji, G.; Wiegman, C.H.; Michaeloudes, C.; Patel, M.S.; Curtis, K.; Bhavsar, P.; Polkey, M.I.; Adcock, I.M.; Chung, K.F.; on behalf of the COPDMAP consortium. Mitochondrial dysfunction in airways and quadriceps muscle of patients with chronic obstructive pulmonary disease. Respir. Res. 2020, 21, 262. [Google Scholar] [CrossRef]
- Zhang, W.Z.; Hoffman, K.L.; Schiffer, K.T.; Oromendia, C.; Rice, M.C.; Barjaktarevic, I.; Peters, S.P.; Putcha, N.; Bowler, R.P.; Wells, J.M.; et al. Association of plasma mitochondrial DNA with COPD severity and progression in the SPIROMICS cohort. Respir. Res. 2021, 22, 126. [Google Scholar] [CrossRef]
- Zhang, W.Z.; Rice, M.C.; Hoffman, K.L.; Oromendia, C.; Barjaktarevic, I.Z.; Wells, J.M.; Hastie, A.T.; Labaki, W.W.; Cooper, C.B.; Comellas, A.P.; et al. Association of urine mitochondrial DNA with clinical measures of COPD in the SPIROMICS cohort. JCI Insight 2020, 5, e133984. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dai, A.; Hu, R.; Zhu, L.; Tan, S. Positive correlation between PPARgamma/PGC-1alpha and gamma-GCS in lungs of rats and patients with chronic obstructive pulmonary disease. Acta Biochim. Biophys. Sin. 2010, 42, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Yang, M.; Chen, Z.Y.; Chen, P.; Guan, C.X.; Xiang, X.D.; Cai, S.; Chen, Y.; Fang, X. Expression and methylation of mitochondrial transcription factor a in chronic obstructive pulmonary disease patients with lung cancer. PLoS ONE 2013, 8, e82739. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Inflammatory endotypes in COPD. Allergy 2019, 74, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Corlateanu, A.; Mendez, Y.; Wang, Y.; Garnica, R.d.J.A.; Botnaru, V.; Siafakas, N. Chronic obstructive pulmonary disease and phenotypes: A state-of-the-art. Pulmonology 2020, 26, 95–100. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: 2022 Report. 2022. Available online: https://goldcopd.org/wp-content/uploads/2021/12/GOLD-REPORT-2022-v1.1-22Nov2021_WMV.pdf (accessed on 23 December 2022).
- Hikichi, M.; Mizumura, K.; Maruoka, S.; Gon, Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J. Thorac. Dis. 2019, 11, S2129–S2140. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef]
- Sundar, I.K.; Maremanda, K.P.; Rahman, I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol. Lett. 2019, 317, 92–101. [Google Scholar] [CrossRef]
- Ballweg, K.; Mutze, K.; Königshoff, M.; Eickelberg, O.; Meiners, S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L895–L907. [Google Scholar] [CrossRef]
- Wu, K.; Luan, G.; Xu, Y.; Shen, S.; Qian, S.; Zhu, Z.; Zhang, X.; Yin, S.; Ye, J. Cigarette smoke extract increases mitochondrial membrane permeability through activation of adenine nucleotide translocator (ANT) in lung epithelial cells. Biochem. Biophys. Res. Commun. 2020, 525, 733–739. [Google Scholar] [CrossRef]
- Van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [Green Version]
- Valdivieso, Á.G.; Dugour, A.V.; Sotomayor, V.; Clauzure, M.; Figueroa, J.M.; Santa-Coloma, T.A. N-acetyl cysteine reverts the proinflammatory state induced by cigarette smoke extract in lung Calu-3 cells. Redox Biol. 2018, 16, 294–302. [Google Scholar] [CrossRef]
- Malinska, D.; Szymanski, J.; Patalas-Krawczyk, P.; Michalska, B.; Wojtala, A.; Prill, M.; Partyka, M.; Drabik, K.; Walczak, J.; Sewer, A.; et al. Assessment of mitochondrial function following short- and long-term exposure of human bronchial epithelial cells to total particulate matter from a candidate modified-risk tobacco product and reference cigarettes. Food Chem. Toxicol. 2018, 115, 1–12. [Google Scholar] [CrossRef]
- Agarwal, A.R.; Yin, F.; Cadenas, E. Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am. J. Respir. Cell Mol. Biol. 2014, 51, 284–293. [Google Scholar] [CrossRef]
- Agarwal, A.R.; Zhao, L.; Sancheti, H.; Sundar, I.K.; Rahman, I.; Cadenas, E. Short-term cigarette smoke exposure induces reversible changes in energy metabolism and cellular redox status independent of inflammatory responses in mouse lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L889–L898. [Google Scholar] [CrossRef] [Green Version]
- Tulen, C.B.M.; Wang, Y.; Beentjes, D.; Jessen, P.J.J.; Ninaber, D.K.; Reynaert, N.L.; van Schooten, F.J.; Opperhuizen, A.; Hiemstra, P.S.; Remels, A.H.V. Dysregulated mitochondrial metabolism upon cigarette smoke exposure in various human bronchial epithelial cell models. Dis. Model. Mech. 2022, 15, dmm049247. [Google Scholar] [CrossRef]
- Vanella, L.; Li Volti, G.; Distefano, A.; Raffaele, M.; Zingales, V.; Avola, R.; Tibullo, D.; Barbagallo, I. A new antioxidant formulation reduces the apoptotic and damaging effect of cigarette smoke extract on human bronchial epithelial cells. Eur. Rev. Med. Pharm. Sci. 2017, 21, 5478–5484. [Google Scholar] [CrossRef]
- Wang, S.; He, N.; Xing, H.; Sun, Y.; Ding, J.; Liu, L. Function of hesperidin alleviating inflammation and oxidative stress responses in COPD mice might be related to SIRT1/PGC-1alpha/NF-kappaB signaling axis. J. Recept. Signal Transduct. Res. 2020, 40, 388–394. [Google Scholar] [CrossRef]
- Wang, X.L.; Li, T.; Li, J.H.; Miao, S.Y.; Xiao, X.Z. The Effects of Resveratrol on Inflammation and Oxidative Stress in a Rat Model of Chronic Obstructive Pulmonary Disease. Molecules 2017, 22, 1529. [Google Scholar] [CrossRef] [Green Version]
- Kyung, S.Y.; Kim, Y.J.; Son, E.S.; Jeong, S.H.; Park, J.W. The Phosphodiesterase 4 Inhibitor Roflumilast Protects against Cigarette Smoke Extract-Induced Mitophagy-Dependent Cell Death in Epithelial Cells. Tuberc. Respir. Dis. 2018, 81, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Son, E.S.; Kim, S.H.; Ryter, S.W.; Yeo, E.J.; Kyung, S.Y.; Kim, Y.J.; Jeong, S.H.; Lee, C.S.; Park, J.W. Quercetogetin protects against cigarette smoke extract-induced apoptosis in epithelial cells by inhibiting mitophagy. Toxicol. Vitr. 2018, 48, 170–178. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, R.; Zhang, Y.; Shan, H.; Zhang, Q.; Yang, X.; Li, Y.; Zhang, J. Nix/BNIP3L-dependent mitophagy accounts for airway epithelial cell injury induced by cigarette smoke. J. Cell. Physiol. 2019, 234, 14210–14220. [Google Scholar] [CrossRef]
- Wen, W.; Yu, G.; Liu, W.; Gu, L.; Chu, J.; Zhou, X.; Liu, Y.; Lai, G. Silencing FUNDC1 alleviates chronic obstructive pulmonary disease by inhibiting mitochondrial autophagy and bronchial epithelium cell apoptosis under hypoxic environment. J. Cell. Biochem. 2019, 120, 17602–17615. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 2019, 15, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Luo, B.; Gong, L. Resveratrol reduces the apoptosis induced by cigarette smoke extract by upregulating MFN2. PLoS ONE 2017, 12, e0175009. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Park, Y.J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol. Lett. 2019, 303, 55–66. [Google Scholar] [CrossRef]
- Walczak, J.; Malińska, D.; Drabik, K.; Michalska, B.; Prill, M.; Johne, S.; Luettich, K.; Szymański, J.; Peitsch, M.C.; Hoeng, J.; et al. Mitochondrial Network and Biogenesis in Response to Short and Long-Term Exposure of Human BEAS-2B Cells to Aerosol Extracts from the Tobacco Heating System 2.2. Cell. Physiol. Biochem. 2020, 54, 230–251. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabon, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef]
- Wang, H.T.; Lin, J.H.; Yang, C.H.; Haung, C.H.; Weng, C.W.; Maan-Yuh Lin, A.; Lo, Y.L.; Chen, W.S.; Tang, M.S. Acrolein induces mtDNA damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget 2017, 8, 70406–70421. [Google Scholar] [CrossRef]
- Tulen, C.B.M.; Snow, S.J.; Leermakers, P.A.; Kodavanti, U.P.; van Schooten, F.J.; Opperhuizen, A.; Remels, A.H.V. Acrolein inhalation acutely affects the regulation of mitochondrial metabolism in rat lung. Toxicology 2022, 469, 153129. [Google Scholar] [CrossRef]
- Fabisiak, J.P.; Medvedovic, M.; Alexander, D.C.; McDunn, J.E.; Concel, V.J.; Bein, K.; Jang, A.S.; Berndt, A.; Vuga, L.J.; Brant, K.A.; et al. Integrative metabolome and transcriptome profiling reveals discordant energetic stress between mouse strains with differential sensitivity to acrolein-induced acute lung injury. Mol. Nutr. Food Res. 2011, 55, 1423–1434. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.R.; Yin, F.; Cadenas, E. Metabolic shift in lung alveolar cell mitochondria following acrolein exposure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L764–L773. [Google Scholar] [CrossRef] [Green Version]
- Roy, J.; Pallepati, P.; Bettaieb, A.; Tanel, A.; Averill-Bates, D.A. Acrolein induces a cellular stress response and triggers mitochondrial apoptosis in A549 cells. Chem. Biol. Interact. 2009, 181, 154–167. [Google Scholar] [CrossRef]
- Luo, C.; Li, Y.; Yang, L.; Feng, Z.; Li, Y.; Long, J.; Liu, J. A cigarette component acrolein induces accelerated senescence in human diploid fibroblast IMR-90 cells. Biogerontology 2013, 14, 503–511. [Google Scholar] [CrossRef]
- Sun, L.; Luo, C.; Long, J.; Wei, D.; Liu, J. Acrolein is a mitochondrial toxin: Effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion 2006, 6, 136–142. [Google Scholar] [CrossRef]
- Sisson, J.H.; Tuma, D.J.; Rennard, S.I. Acetaldehyde-mediated cilia dysfunction in bovine bronchial epithelial cells. Am. J. Physiol. 1991, 260, L29–L36. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Schmidt, S.C.; Rennard, S.I.; Tuma, D.J.; Sisson, J.H. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc. Soc. Exp. Biol. Med. 2000, 225, 91–97. [Google Scholar] [CrossRef]
- Farfán Labonne, B.E.; Gutiérrez, M.; Gómez-Quiroz, L.E.; Konigsberg Fainstein, M.; Bucio, L.; Souza, V.; Flores, O.; Ortíz, V.; Hernández, E.; Kershenobich, D.; et al. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol. Toxicol. 2009, 25, 599–609. [Google Scholar] [CrossRef]
- Van Buskirk, J.J.; Frisell, W.R. Inhibition by formaldehyde of energy transfer and related processes in rat liver mitochondria. II. Effects on energy-linked reactions in intact mitochondria and phosphorylating particles. Arch. Biochem. Biophys 1969, 132, 130–138. [Google Scholar] [CrossRef]
- Zerin, T.; Kim, J.S.; Gil, H.W.; Song, H.Y.; Hong, S.Y. Effects of formaldehyde on mitochondrial dysfunction and apoptosis in SK-N-SH neuroblastoma cells. Cell Biol. Toxicol. 2015, 31, 261–272. [Google Scholar] [CrossRef]
- Ma, H.; Lin, J.; Li, L.; Ding, Z.; Huang, P.; Song, X.; Lou, K.; Wang, W.; Xu, H. Formaldehyde reinforces pro-inflammatory responses of macrophages through induction of glycolysis. Chemosphere 2021, 282, 131149. [Google Scholar] [CrossRef]
- Li, S.; Wei, P.; Zhang, B.; Chen, K.; Shi, G.; Zhang, Z.; Du, Z. Apoptosis of lung cells regulated by mitochondrial signal pathway in crotonaldehyde-induced lung injury. Environ. Toxicol. 2020, 35, 1260–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, B.; Zhang, Q.; Zhang, Z. Crotonaldehyde exposure induces liver dysfunction and mitochondrial energy metabolism disorder in rats. Toxicol. Mech. Methods 2021, 31, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Yang, Z.H.; Pan, X.J.; Zhu, M.X.; Xie, J.P. Crotonaldehyde induces oxidative stress and caspase-dependent apoptosis in human bronchial epithelial cells. Toxicol. Lett. 2010, 195, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.C.; Pan, X.J.; Yang, Z.H.; Xiao, F.J.; Liu, X.Y.; Zhu, M.X.; Xie, J.P. Crotonaldehyde induces apoptosis in alveolar macrophages through intracellular calcium, mitochondria and p53 signaling pathways. J. Toxicol. Sci. 2013, 38, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, Z.; Zhuang, Z.; Sang, H.; Wu, Z.; Meng, R.; He, E.Y.; Scott, G.I.; Maris, J.R.; Li, R.; Ren, J. α,β-Unsaturated aldehyde crotonaldehyde triggers cardiomyocyte contractile dysfunction: Role of TRPV1 and mitochondrial function. Pharm. Res. 2014, 82, 40–50. [Google Scholar] [CrossRef]
- Clapp, P.W.; Lavrich, K.S.; van Heusden, C.A.; Lazarowski, E.R.; Carson, J.L.; Jaspers, I. Cinnamaldehyde in flavored e-cigarette liquids temporarily suppresses bronchial epithelial cell ciliary motility by dysregulation of mitochondrial function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L470–L486. [Google Scholar] [CrossRef]
- Liu, Q.P.; Zhou, D.X.; Lv, M.Q.; Ge, P.; Li, Y.X.; Wang, S.J. Formaldehyde inhalation triggers autophagy in rat lung tissues. Toxicol. Ind. Health 2018, 34, 834–841. [Google Scholar] [CrossRef]
- Caldeira, D.A.F.; Weiss, D.J.; Rocco, P.R.M.; Silva, P.L.; Cruz, F.F. Mitochondria in Focus: From Function to Therapeutic Strategies in Chronic Lung Diseases. Front. Immunol. 2021, 12, 782074. [Google Scholar] [CrossRef]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Mabalirajan, U. Rejuvenating cellular respiration for optimizing respiratory function: Targeting mitochondria. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L103–L113. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.-H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Perez-Miller, S.; Younus, H.; Vanam, R.; Chen, C.H.; Mochly-Rosen, D.; Hurley, T.D. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat. Struct. Mol. Biol. 2010, 17, 159–164. [Google Scholar] [CrossRef]
- Kim, C.; Hwang, S.; Choi, H.J.; Lim, T.H.; Kang, J.-S. The Effect of Aldehyde Dehydrogenase Activator, Alda-1®, on the Ethanol-induced Brain Damage in a Rat of Binge Ethanol Intoxication. bioRxiv 2018, 353854. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.-H.; Zhang, H.-F.; Yang, Z.-B.; Li, T.-B.; Liu, B.; Lou, Z.; Ma, Q.-L.; Luo, X.-J.; Peng, J. Alda-1 reduces cerebral ischemia/reperfusion injury in rat through clearance of reactive aldehydes. Naunyn-Schmiedebergs Arch. Pharmacol. 2014, 387, 87–94. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Yeh, T.-H.; Lai, S.-C.; Wu-Chou, Y.-H.; Chen, C.-H.; Mochly-Rosen, D.; Huang, Y.-C.; Chen, Y.-J.; Chen, C.-L.; Chang, Y.-M.; et al. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp. Neurol. 2015, 263, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Mundy, M.; Chambers, E.; Lange, T.; Newton, J.; Borgas, D.; Yao, H.; Choudhary, G.; Basak, R.; Oldham, M.; et al. Alda-1 Protects Against Acrolein-Induced Acute Lung Injury and Endothelial Barrier Dysfunction. Am. J. Respir. Cell Mol. Biol. 2017, 57, 662–673. [Google Scholar] [CrossRef]
- Patil, S.S.; Hernández-Cuervo, H.; Fukumoto, J.; Narala, V.R.; Saji, S.; Borra, M.; Alleyn, M.; Lin, M.; Soundararajan, R.; Lockey, R.; et al. Alda-1 attenuates hyperoxia-induced mitochondrial dysfunction in lung vascular endothelial cells. Aging 2019, 11, 3909–3918. [Google Scholar] [CrossRef]
- Patil, S.S.; Hernández-Cuervo, H.; Fukumoto, J.; Krishnamurthy, S.; Lin, M.; Alleyn, M.; Breitzig, M.; Narala, V.R.; Soundararajan, R.; Lockey, R.F.; et al. Alda-1 Attenuates Hyperoxia-Induced Acute Lung Injury in Mice. Front. Pharm. 2020, 11, 597942. [Google Scholar] [CrossRef]
- Tsai, H.Y.; Hsu, Y.J.; Lu, C.Y.; Tsai, M.C.; Hung, W.C.; Chen, P.C.; Wang, J.C.; Hsu, L.A.; Yeh, Y.H.; Chu, P.; et al. Pharmacological Activation Of Aldehyde Dehydrogenase 2 Protects Against Heatstroke-Induced Acute Lung Injury by Modulating Oxidative Stress and Endothelial Dysfunction. Front. Immunol. 2021, 12, 740562. [Google Scholar] [CrossRef]
- Cao, Z.; Qin, H.; Huang, Y.; Zhao, Y.; Chen, Z.; Hu, J.; Gao, Q. Crosstalk of pyroptosis, ferroptosis, and mitochondrial aldehyde dehydrogenase 2-related mechanisms in sepsis-induced lung injury in a mouse model. Bioengineered 2022, 13, 4810–4820. [Google Scholar] [CrossRef]
- Li, K.; Guo, W.; Li, Z.; Wang, Y.; Sun, B.; Xu, D.; Ling, J.; Song, H.; Liao, Y.; Wang, T.; et al. ALDH2 Repression Promotes Lung Tumor Progression via Accumulated Acetaldehyde and DNA Damage. Neoplasia 2019, 21, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Yang, M.; Zhou, Y.; Chen, L.; Wu, H.; Liu, R. Alda-1 Prevents Pulmonary Epithelial Barrier Dysfunction following Severe Hemorrhagic Shock through Clearance of Reactive Aldehydes. Biomed. Res. Int. 2019, 2019, 2476252. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Luo, Q.; Zhu, H.; Liu, X.; Dong, Z.; Zhang, K.; Zou, Y.; Wu, J.; Ge, J.; Sun, A. Aldehyde dehydrogenase 2 activation ameliorates CCl(4) -induced chronic liver fibrosis in mice by up-regulating Nrf2/HO-1 antioxidant pathway. J. Cell. Mol. Med. 2018, 22, 3965–3978. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Xing, J.H.; Zhang, C.; Zhang, Y.M.; Zhang, L.T.; Wei, S.J.; Zhang, M.X.; Wang, X.P.; Yuan, Q.H.; Xue, L.; et al. Aldehyde dehydrogenase 2 inhibits inflammatory response and regulates atherosclerotic plaque. Oncotarget 2016, 7, 35562–35576. [Google Scholar] [CrossRef] [Green Version]
- Panisello-Roselló, A.; Lopez, A.; Folch-Puy, E.; Carbonell, T.; Rolo, A.; Palmeira, C.; Adam, R.; Net, M.; Roselló-Catafau, J. Role of aldehyde dehydrogenase 2 in ischemia reperfusion injury: An update. World J. Gastroenterol. 2018, 24, 2984–2994. [Google Scholar] [CrossRef]
- Burcham, P.C.; Pyke, S.M. Hydralazine inhibits rapid acrolein-induced protein oligomerization: Role of aldehyde scavenging and adduct trapping in cross-link blocking and cytoprotection. Mol. Pharm. 2006, 69, 1056–1065. [Google Scholar] [CrossRef] [Green Version]
- Wondrak, G.T.; Cervantes-Laurean, D.; Roberts, M.J.; Qasem, J.G.; Kim, M.; Jacobson, E.L.; Jacobson, M.K. Identification of alpha-dicarbonyl scavengers for cellular protection against carbonyl stress. Biochem. Pharm. 2002, 63, 361–373. [Google Scholar] [CrossRef]
- Vistoli, G.; Orioli, M.; Pedretti, A.; Regazzoni, L.; Canevotti, R.; Negrisoli, G.; Carini, M.; Aldini, G. Design, synthesis, and evaluation of carnosine derivatives as selective and efficient sequestering agents of cytotoxic reactive carbonyl species. ChemMedChem 2009, 4, 967–975. [Google Scholar] [CrossRef]
- Rahman, I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim. Biophys. Acta 2012, 1822, 714–728. [Google Scholar] [CrossRef] [Green Version]
- Moretti, M.; Bottrighi, P.; Dallari, R.; Da Porto, R.; Dolcetti, A.; Grandi, P.; Garuti, G.; Guffanti, E.; Roversi, P.; De Gugliemo, M.; et al. The effect of long-term treatment with erdosteine on chronic obstructive pulmonary disease: The EQUALIFE Study. Drugs Exp. Clin. Res. 2004, 30, 143–152. [Google Scholar]
- Dal Negro, R.; Visconti, M.; Tognella, S.; Micheletto, C. Erdosteine affects eicosanoid production in COPD. Int. J. Clin. Pharmacol. Ther. 2011, 49, 41–45. [Google Scholar] [CrossRef]
- Dal Negro, R.; Visconti, M.; Micheletto, C.; Tognella, S. Erdosteine 900 mg/day leads to substantial changes in blood ROS, e-NO and some chemotactic cytokines in human secretions of current smokers. Am. J. Respir. Crit. Care Med. Suppl. 2005, 2, A89. [Google Scholar]
- Dal Negro, R.W.; Visconti, M.; Micheletto, C.; Tognella, S. Changes in blood ROS, e-NO, and some pro-inflammatory mediators in bronchial secretions following erdosteine or placebo: A controlled study in current smokers with mild COPD. Pulm. Pharm. Ther. 2008, 21, 304–308. [Google Scholar] [CrossRef]
- Tian, R.; Shi, R. Dimercaprol is an acrolein scavenger that mitigates acrolein-mediated PC-12 cells toxicity and reduces acrolein in rat following spinal cord injury. J. Neurochem. 2017, 141, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Lin, Y.; Jiao, Y.; Herr, S.A.; Tang, J.; Rogers, E.; Chen, Z.; Shi, R. Acrolein scavenger dimercaprol offers neuroprotection in an animal model of Parkinson’s disease: Implication of acrolein and TRPA1. Transl. Neurodegener. 2021, 10, 13. [Google Scholar] [CrossRef]
- Mai, X.; Zhou, F.; Lin, P.; Lin, S.; Gao, J.; Ma, Y.; Fan, R.; Ting, W.; Huang, C.; Yin, D.; et al. Metformin scavenges formaldehyde and attenuates formaldehyde-induced bovine serum albumin crosslinking and cellular DNA damage. Environ. Toxicol. 2020, 35, 1170–1178. [Google Scholar] [CrossRef]
- Baker, R.R.; da Silva, J.R.P.; Smith, G. The effect of tobacco ingredients on smoke chemistry. Part I: Flavourings and additives. Food Chem. Toxicol. 2004, 42, 3–37. [Google Scholar] [CrossRef]
- Pauwels, C.; Boots, A.W.; Visser, W.F.; Pennings, J.L.A.; Talhout, R.; Schooten, F.V.; Opperhuizen, A. Characteristic Human Individual Puffing Profiles Can Generate More TNCO than ISO and Health Canada Regimes on Smoking Machine When the Same Brand Is Smoked. Int J. Environ. Res. Public Health 2020, 17, 3225. [Google Scholar] [CrossRef]
- Sthijns, M.M.J.P.E.; Randall, M.J.; Bast, A.; Haenen, G.R.M.M. Adaptation to acrolein through upregulating the protection by glutathione in human bronchial epithelial cells: The materialization of the hormesis concept. Biochem. Biophys. Res. Commun. 2014, 446, 1029–1034. [Google Scholar] [CrossRef]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular mechanisms of acrolein toxicity: Relevance to human disease. Toxicol. Sci. 2015, 143, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Hirata, N.; Yamada, S.; Sekino, Y.; Kanda, Y. Tobacco nitrosamine NNK increases ALDH-positive cells via ROS-Wnt signaling pathway in A549 human lung cancer cells. J. Toxicol. Sci. 2017, 42, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churg, A.; Cosio, M.; Wright, J.L. Mechanisms of cigarette smoke-induced COPD: Insights from animal models. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L612–L631. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Sin, D.D.; Wright, J.L. Everything prevents emphysema: Are animal models of cigarette smoke-induced chronic obstructive pulmonary disease any use? Am. J. Respir. Cell Mol. Biol. 2011, 45, 1111–1115. [Google Scholar] [CrossRef]
- Churg, A.; Wright, J.L. Testing drugs in animal models of cigarette smoke-induced chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 550–552. [Google Scholar] [CrossRef]
- Iskandar, A.R.; Xiang, Y.; Frentzel, S.; Talikka, M.; Leroy, P.; Kuehn, D.; Guedj, E.; Martin, F.; Mathis, C.; Ivanov, N.V.; et al. Impact Assessment of Cigarette Smoke Exposure on Organotypic Bronchial Epithelial Tissue Cultures: A Comparison of Mono-Culture and Coculture Model Containing Fibroblasts. Toxicol. Sci. 2015, 147, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Van Riet, S.; van Schadewijk, A.; de Vos, S.; Vandeghinste, N.; Rottier, R.J.; Stolk, J.; Hiemstra, P.S.; Khedoe, P. Modulation of Airway Epithelial Innate Immunity and Wound Repair by M(GM-CSF) and M(M-CSF) Macrophages. J. Innate Immun. 2020, 12, 410–421. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tulen, C.B.M.; Opperhuizen, A.; van Schooten, F.-J.; Remels, A.H.V. Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit? Cells 2023, 12, 299. https://doi.org/10.3390/cells12020299
Tulen CBM, Opperhuizen A, van Schooten F-J, Remels AHV. Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit? Cells. 2023; 12(2):299. https://doi.org/10.3390/cells12020299
Chicago/Turabian StyleTulen, Christy B. M., Antoon Opperhuizen, Frederik-Jan van Schooten, and Alexander H. V. Remels. 2023. "Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit?" Cells 12, no. 2: 299. https://doi.org/10.3390/cells12020299
APA StyleTulen, C. B. M., Opperhuizen, A., van Schooten, F. -J., & Remels, A. H. V. (2023). Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit? Cells, 12(2), 299. https://doi.org/10.3390/cells12020299