
Characterization of Hepatoma-Derived Growth Factor-Related Protein 2 Interactions with Heterochromatin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. shRNA Construction
2.3. Lentiviral Transduction
2.4. shRNA Knockdown
2.5. RNA Extraction and qRT-PCR
2.6. Flow Cytometry
2.7. Generation and Validation of CRISPR/Cas9-Mediated HRP2 Knockout Lines
2.8. Co-Immunoprecipitation and Western Blot Analysis
2.9. Immunofluorescence Microscopy
2.10. Sucrose Density Gradient Ultracentrifugation and Fractionation
2.11. Chromatin Immunoprecipitation (ChIP) and qPCR
2.12. Antibodies
3. Results
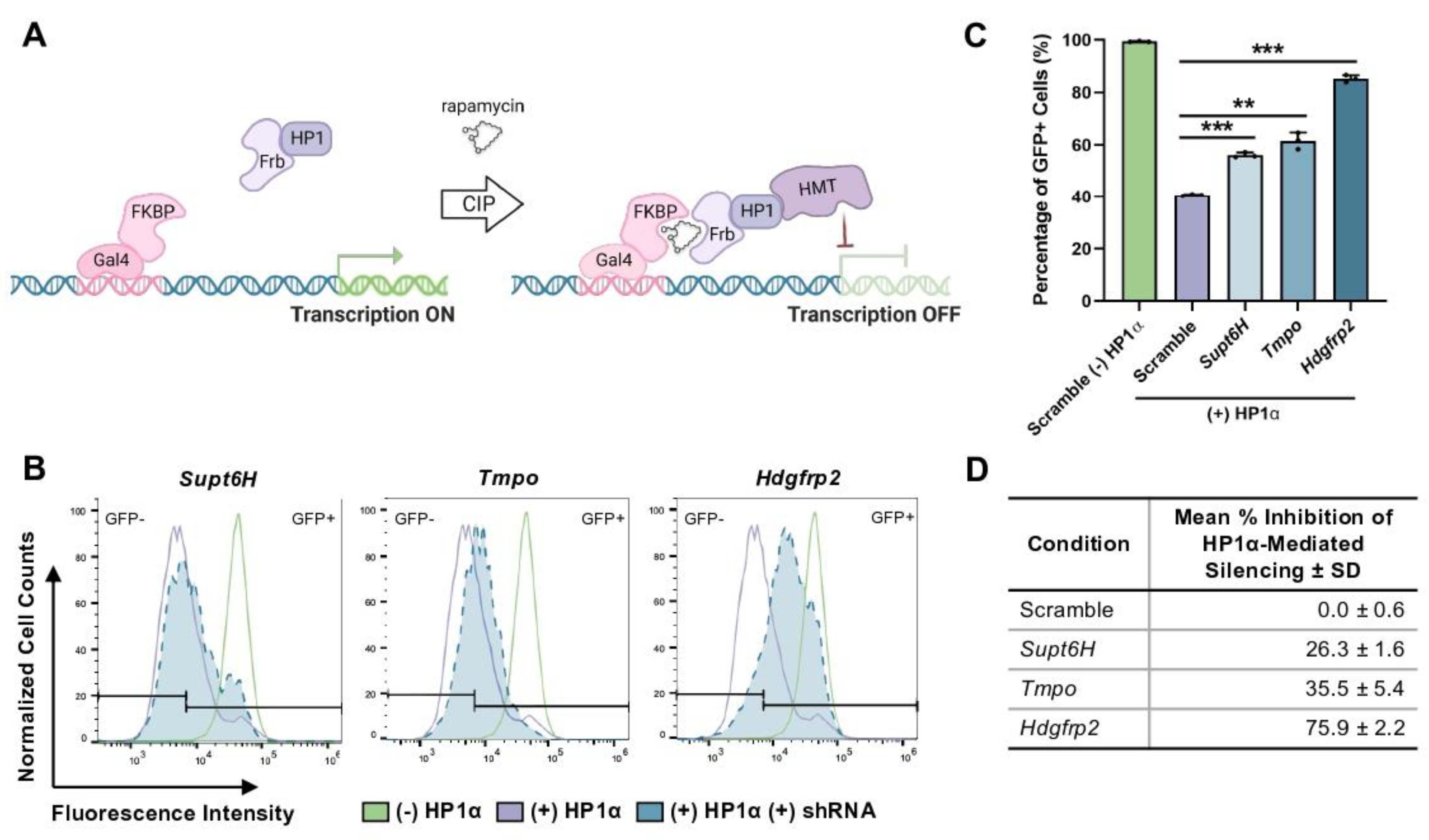
3.1. Hdgfrp2 Knockdown Inhibits HP1α-Mediated Gene Repression
3.2. HRP2 Interacts and Co-Migrates with H3K9me3-Depositing Complexes
3.3. Loss of HRP2 Disrupts HP1α-Mediated Heterochromatin Stability
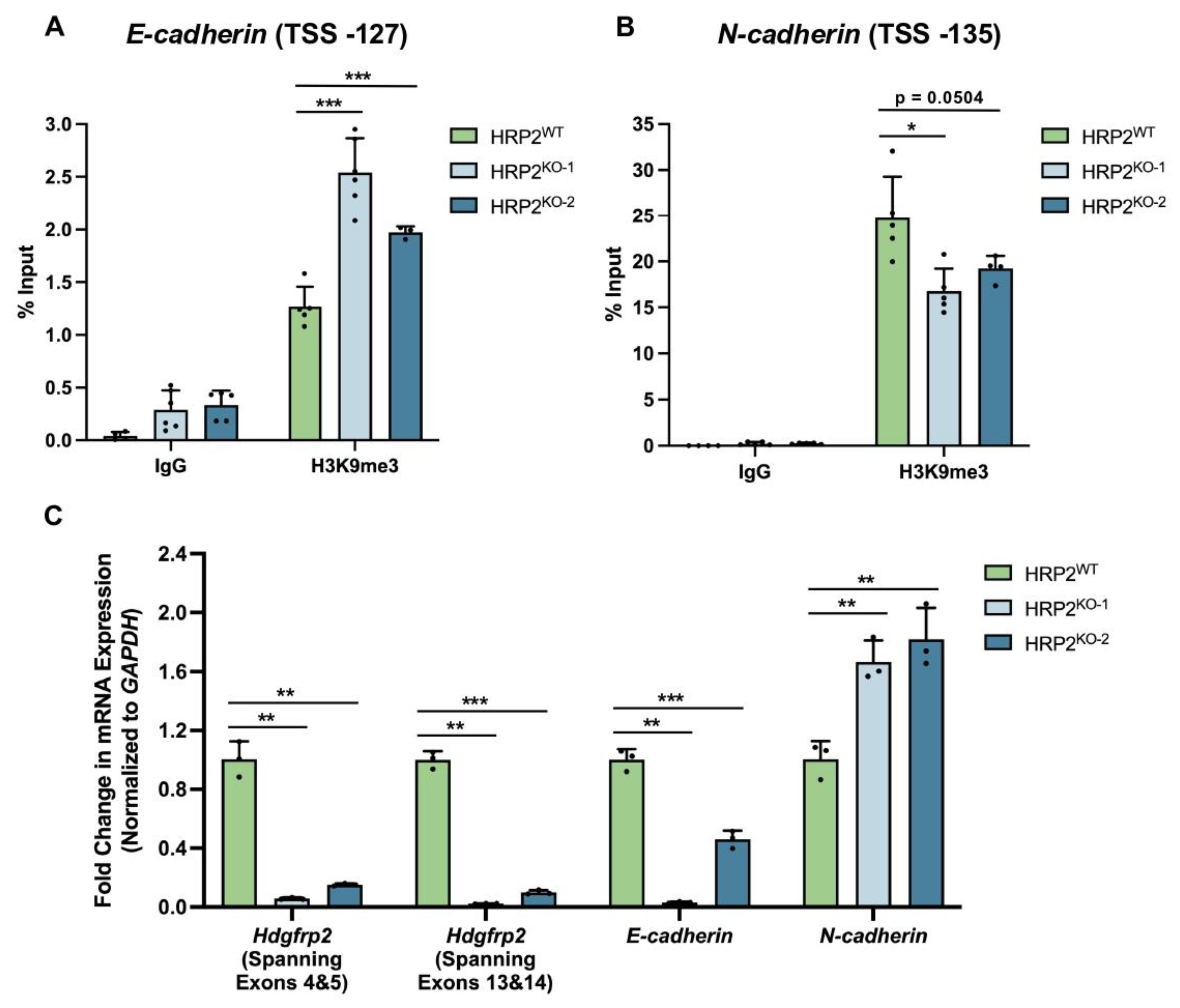
3.4. HRP2 Knockout Impacts H3K9me3 Occupancy at E- and N-Cadherin, Resulting in Altered Transcriptional Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Minc, E.; Allory, Y.; Worman, H.J.; Courvalin, J.C.; Buendia, B. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 1999, 108, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol. Cell 2005, 19, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.A.; Taverna, S.D.; Zhang, Y.; Briggs, S.D.; Li, J.; Eissenberg, J.C.; Allis, C.D.; Khorasanizadeh, S. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 2001, 20, 5232. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, P.R.; Nietlispach, D.; Mott, H.R.; Callaghan, J.; Bannister, A.; Kouzarides, T.; Murzin, A.G.; Murzina, N.V.; Laue, E.D. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 2002, 416, 103–107. [Google Scholar] [CrossRef]
- Smothers, J.F.; Henikoff, S. The Hinge and Chromo Shadow Domain Impart Distinct Targeting of HP1-Like Proteins. Mol. Cell. Biol. 2001, 21, 2555–2569. [Google Scholar] [CrossRef] [Green Version]
- Kilic, S.; Bachmann, A.L.; Bryan, L.C.; Fierz, B. Multivalency governs HP1α association dynamics with the silent chromatin state. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.F.M.; Mermoud, J.E.; O’carroll, D.; Pagani, M.; Schweizer, D.; Brockdorff, N.; Jenuwein, T. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 2002, 30, 77–80. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Matheson, N.J.; Wals, K.; Antrobus, R.; Göttgens, B.; Dougan, G.; Dawson, M.A.; Lehner, P.J. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 2015, 348, 1481–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, Z.; Ruan, J.; Xu, C.; Tong, Y.; Pan, P.W.; Tempel, W.; Crombet, L.; Min, J.; Zang, J. Structural basis for specific binding of human MPP8 chromodomain to histone H3 methylated at lysine 9. PLoS ONE 2011, 6, e25104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Sun, L.; Kokura, K.; Horton, J.R.; Fukuda, M.; Espejo, A.; Izumi, V.; Koomen, J.M.; Bedford, M.T.; Zhang, X.; et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011, 2, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Tapias, P.; Robin, P.; Pontis, J.; Del Maestro, L.; Ait-Si-Ali, S. The H3K9 Methylation Writer SETDB1 and its Reader MPP8 Cooperate to Silence Satellite DNA Repeats in Mouse Embryonic Stem Cells. Genes 2019, 10, 750. [Google Scholar] [CrossRef] [Green Version]
- Müller, I.; Moroni, A.S.; Shlyueva, D.; Sahadevan, S.; Schoof, E.M.; Radzisheuskaya, A.; Højfeldt, J.W.; Tatar, T.; Koche, R.P.; Huang, C.; et al. MPP8 is essential for sustaining self-renewal of ground-state pluripotent stem cells. Nat. Commun. 2021, 12, 3034. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C. Stop HUSHing on SIV/HIV. Nat. Microbiol. 2018, 3, 1336–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, I.A.; Butler, K.V.; Herring, L.E.; Clinkscales, S.E.; Yelagandula, R.; Stecher, K.; Bell, O.; Graves, L.M.; Jin, J.; Hathaway, N.A. Pathway-Based High-Throughput Chemical Screen Identifies Compounds That Decouple Heterochromatin Transformations. SLAS Discov. 2019, 24, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.H.; Zare, H.; Mousavi, K.; Wang, C.; Moravec, C.E.; Sirotkin, H.I.; Ge, K.; Gutierrez-Cruz, G.; Sartorelli, V. The histone chaperone Spt6 coordinates histone H3K27 demethylation and myogenesis. EMBO J. 2013, 32, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narain, A.; Bhandare, P.; Adhikari, B.; Backes, S.; Eilers, M.; Dölken, L.; Schlosser, A.; Erhard, F.; Baluapuri, A.; Wolf, E. Targeted protein degradation reveals a direct role of SPT6 in RNAPII elongation and termination. Mol. Cell 2021, 81, 3110. [Google Scholar] [CrossRef]
- Wang, H.; Jurado, K.A.; Wu, X.; Shun, M.C.; Li, X.; Ferris, A.L.; Smith, S.J.; Patel, P.A.; Fuchs, J.R.; Cherepanov, P.; et al. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012, 40, 11518–11530. [Google Scholar] [CrossRef]
- Schrijvers, R.; Vets, S.; De Rijck, J.; Malani, N.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 2012, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zeng, H.; Lam, R.; Tempel, W.; Amaya, M.F.; Xu, C.; Dombrovski, L.; Qiu, W.; Wang, Y.; Min, J. Structural and Histone Binding Ability Characterizations of Human PWWP Domains. PLoS ONE 2011, 6, e18919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoy, G.; Oksuz, O.; Descostes, N.; Aoi, Y.; Ganai, R.A.; Kara, H.O.; Yu, J.R.; Lee, C.H.; Stafford, J.; Shilatifard, A.; et al. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci. Adv. 2019, 5, eaay3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Lan, B.; Yi, X.; He, C.; Dang, L.; Zhou, X.; Lu, Y.; Sun, Y.; Liu, Z.; Bai, X.; et al. HRP2-DPF3a-BAF complex coordinates histone modification and chromatin remodeling to regulate myogenic gene transcription. Nucleic Acids Res. 2020, 48, 6563–6582. [Google Scholar] [CrossRef] [PubMed]
- Baude, A.; Aaes, T.L.; Zhai, B.; Al-Nakouzi, N.; Oo, H.Z.; Daugaard, M.; Rohde, M.; Jäättelä, M. Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res. 2015, 44, 2214–2226. [Google Scholar] [CrossRef] [Green Version]
- Waybright, J.M.; Clinkscales, S.E.; Barnash, K.D.; Budziszewski, G.R.; Rectenwald, J.M.; Chiarella, A.M.; Norris-Drouin, J.L.; Cholensky, S.H.; Pearce, K.H.; Herring, L.E.; et al. A Peptidomimetic Ligand Targeting the Chromodomain of MPP8 Reveals HRP2′s Association with the HUSH Complex. ACS Chem. Biol. 2021, 16, 1721–1736. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, X.; Dang, L.; Jiang, H.; Xie, Y.; Li, X.; Guo, J.; Wang, Y.; Peng, Z.; Wang, M.; et al. Epigenomic reprogramming via HRP2-MINA dictates response to proteasome inhibitors in multiple myeloma with t(4;14) translocation. J. Clin. Investig. 2022, 132, e149526. [Google Scholar] [CrossRef] [PubMed]
- Kokura, K.; Sun, L.; Bedford, M.T.; Fang, J. Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J. 2010, 29, 3673. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is Critical for Snail-mediated E-cadherin Repression in Breast Cancer. Oncogene 2013, 32, 1351. [Google Scholar] [CrossRef] [Green Version]
- Hathaway, N.A.; Bell, O.; Hodges, C.; Miller, E.L.; Neel, D.S.; Crabtree, G.R. Dynamics and memory of heterochromatin in living cells. Cell 2012, 149, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Foley, C.A.; Birla, S.V.; Hepperla, A.J.; Simon, J.M.; James, L.I.; Hathaway, N.A. Bioorthogonal Chemical Epigenetic Modifiers Enable Dose-Dependent CRISPR Targeted Gene Activation in Mammalian Cells. ACS Synth. Biol. 2022, 11, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Feldman, N.; Gerson, A.; Fang, J.; Li, E.; Zhang, Y.; Shinkai, Y.; Cedar, H.; Bergman, Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell Biol. 2006, 8, 188–194. [Google Scholar] [CrossRef]
- Lu, P.; Hostager, B.S.; Rothman, P.B.; Colgan, J.D. Sedimentation and Immunoprecipitation Assays for Analyzing Complexes that Repress Transcription. Methods Mol. Biol. 2013, 977, 365. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Martinez, J.; LaCava, J.; Rout, M.P. Density Gradient Ultracentrifugation to Isolate Endogenous Protein Complexes after Affinity Capture. Cold Spring Harb. Protoc. 2016, 2016, 624–627. [Google Scholar] [CrossRef]
- Moon, K.J.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Prigozhin, D.M.; Douse, C.H.; Farleigh, L.E.; Albecka, A.; Tchasovnikarova, I.A.; Timms, R.T.; Oda, S.I.; Adolf, F.; Freund, S.M.V.; Maslen, S.; et al. Periphilin self-association underpins epigenetic silencing by the HUSH complex. Nucleic Acids Res. 2020, 48, 10313–10328. [Google Scholar] [CrossRef] [PubMed]
- Bierer, B.E.; Mattila, P.S.; Standaert, R.F.; Herzenberg, L.A.; Burakoff, S.J.; Crabtree, G.; Schreiber, S.L. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. USA 1990, 87, 9231–9235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Kokura, K.; Izumi, V.; Koomen, J.M.; Seto, E.; Chen, J.; Fang, J. MPP8 and SIRT1 crosstalk in E-cadherin gene silencing and epithelial-mesenchymal transition. EMBO Rep. 2015, 16, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Yu, P.; Tang, J. Characterization of Triple-Negative Breast Cancer MDA-MB-231 Cell Spheroid Model. Onco. Targets Ther. 2020, 13, 5395. [Google Scholar] [CrossRef] [PubMed]
- Izumoto, Y.; Kuroda, T.; Harada, H.; Kishimoto, T.; Nakamura, H. Hepatoma-Derived Growth Factor Belongs to a Gene Family in Mice Showing Significant Homology in the Amino Terminus. Biochem. Biophys. Res. Commun. 1997, 238, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Everett, A.D. Hepatoma derived growth factor binds DNA through the N-terminal PWWP domain. BMC Mol. Biol. 2007, 8, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, Y.L.; Lee, H.J.; Jiang, I.; Lin, S.C.; Lo, W.C.; Lin, Y.J.; Sue, S.C. The First Residue of the PWWP Motif Modulates HATH Domain Binding, Stability, and Protein-Protein Interaction. Biochemistry 2015, 54, 4063–4074. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.; Bensi, D.; Fernandez-Zapico, M.E.; Urrutia, R. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat. Cell Biol. 2006, 8, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Abmayr, S.M.; Workman, J.L. Heterochromatin protein 1 (HP1) connects the FACT histone chaperone complex to the phosphorylated CTD of RNA polymerase II. Genes Dev. 2010, 24, 2133–2145. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wistner, S.C.; MacDonald, I.A.; Stanley, K.A.; Hathaway, N.A. Characterization of Hepatoma-Derived Growth Factor-Related Protein 2 Interactions with Heterochromatin. Cells 2023, 12, 325. https://doi.org/10.3390/cells12020325
Wistner SC, MacDonald IA, Stanley KA, Hathaway NA. Characterization of Hepatoma-Derived Growth Factor-Related Protein 2 Interactions with Heterochromatin. Cells. 2023; 12(2):325. https://doi.org/10.3390/cells12020325
Chicago/Turabian StyleWistner, Sarah C., Ian A. MacDonald, Karly A. Stanley, and Nathaniel A. Hathaway. 2023. "Characterization of Hepatoma-Derived Growth Factor-Related Protein 2 Interactions with Heterochromatin" Cells 12, no. 2: 325. https://doi.org/10.3390/cells12020325
APA StyleWistner, S. C., MacDonald, I. A., Stanley, K. A., & Hathaway, N. A. (2023). Characterization of Hepatoma-Derived Growth Factor-Related Protein 2 Interactions with Heterochromatin. Cells, 12(2), 325. https://doi.org/10.3390/cells12020325