
Examining the Role of a Functional Deficiency of Iron in Lysosomal Storage Disorders with Translational Relevance to Alzheimer’s Disease

Abstract
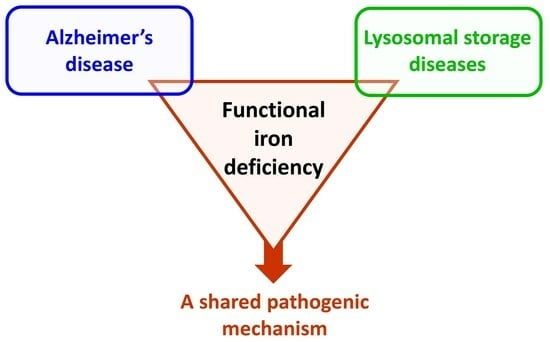
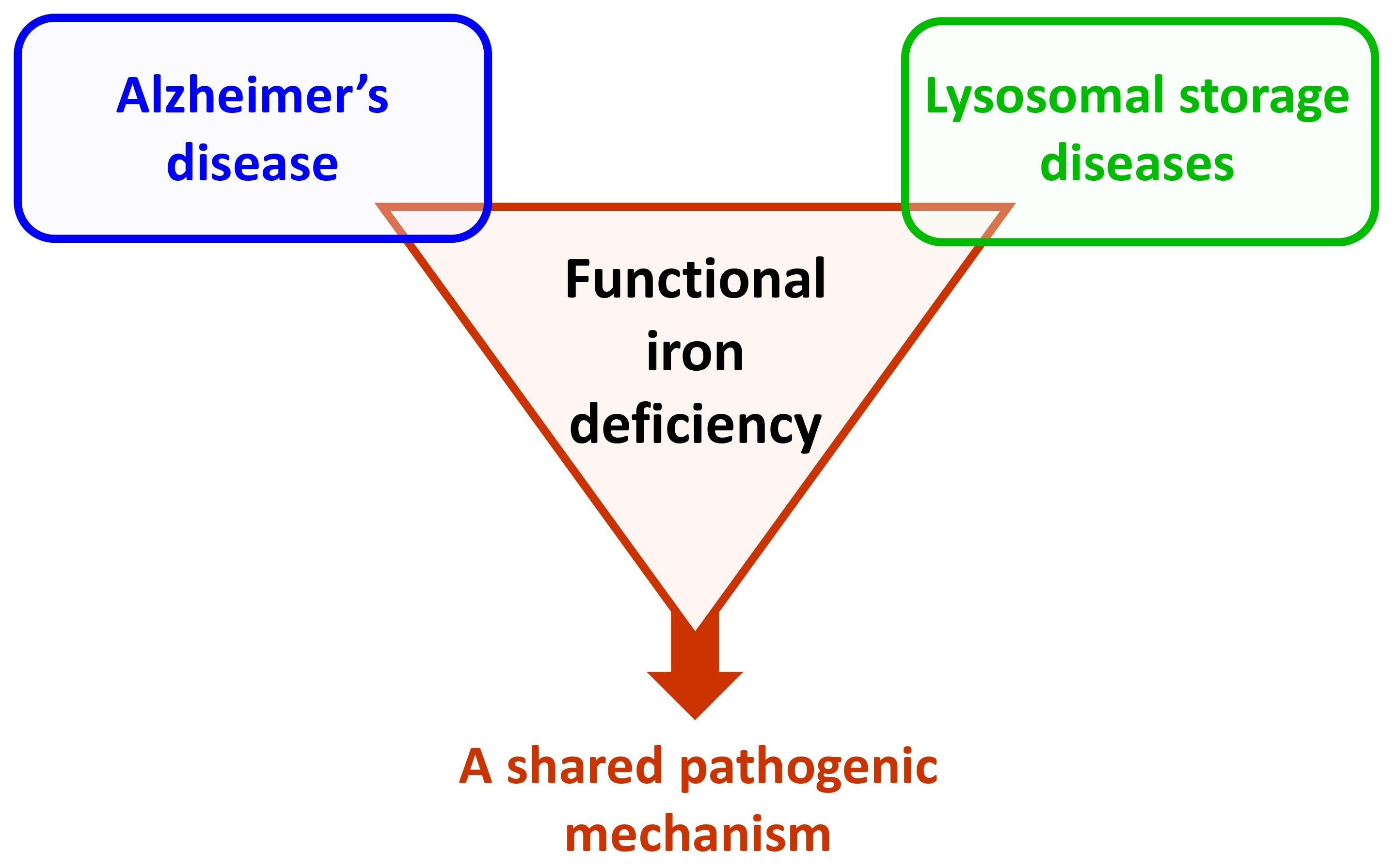
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Features of a Functional Iron Deficiency
3. Lysosomal Storage Diseases and Iron
3.1. Overview of Iron Perturbations in Lysosomal Storage Diseases
3.2. Mucolipidosis Type IV—Highlight of Several Pathological Features
3.3. Mucolipidosis Type IV Causes the Dysfunction of TRPML1, a Lysosomal Channel for Cations Including Fe2+
3.4. Mucolipidosis Type IV Impairs Myelination
3.5. The Potential Impact of a Functional Iron Deficiency in Mucolipidosis Type IV
3.6. Niemann–Pick Type C Disease, Gangliosides, and GM2 Gangliosidosis
3.7. Disturbances to Iron Homeostasis in Niemann–Pick Type C Disease
3.8. Alterations of Iron Homeostasis in Neuronal Ceroid Lipofuscinosis
3.9. Gaucher’s Disease—A Deficiency of Glucosylceramidase
3.9.1. Gaucher’s Disease, α-Synuclein, Mitochondria, and Iron
3.9.2. Gaucher’s Disease, Anemia, and Brain Development
3.9.3. Gaucher’s Disease and a Functional Deficiency of Iron
3.10. Similarities between Krabbe’s Disease and Gaucher’s Disease
3.11. Krabbe’s Disease Models and Iron
3.12. The Likelihood of a Functional Iron Deficiency in Gaucher’s or Krabbe’s Disease
4. Iron in Alzheimer’s Disease and Lysosomal Storage Diseases
4.1. Baseline Characteristics of Lysosomal Storage Disorders and Alzheimer’s Disease Relative to a Functional Iron Deficiency
4.2. Comparing the Pathways Contributing to a Functional Iron Deficiency in Lysosomal Storage Diseases and Alzheimer’s Disease
4.3. Shared Consequences of a Functional Iron Deficiency in Lysosomal Storage Diseases and Alzheimer’s Disease
5. Concluding Thoughts
Funding
Conflicts of Interest
References
- LeVine, S.M. The Azalea Hypothesis of Alzheimer Disease: A Functional Iron Deficiency Promotes Neurodegeneration. Neuroscientist 2023, 10738584231191743. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Ghilardi, J.R.; Rogers, S.; DeMaster, E.; Allen, C.J.; Stimson, E.R.; Maggio, J.E. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of β-amyloid peptide. J. Neurochem. 1993, 61, 1171–1174. [Google Scholar] [CrossRef]
- Kostka, M.; Högen, T.; Danzer, K.M.; Levin, J.; Habeck, M.; Wirth, A.; Wagner, R.; Glabe, C.G.; Finger, S.; Heinzelmann, U.; et al. Single particle characterization of iron-induced pore-forming α-synuclein oligomers. J. Biol. Chem. 2008, 283, 10992–11003. [Google Scholar] [CrossRef]
- Bader, B.; Nübling, G.; Mehle, A.; Nobile, S.; Kretzschmar, H.; Giese, A. Single particle analysis of tau oligomer formation induced by metal ions and organic solvents. Biochem. Biophys. Res. Commun. 2011, 411, 190–196. [Google Scholar] [CrossRef]
- Ahmadi, S.; Ebralidze, I.I.; She, Z.; Kraatz, H.-B. Electrochemical studies of tau protein-iron interactions—Potential implications for Alzheimer’s Disease. Electrochim. Acta 2017, 236, 384–393. [Google Scholar] [CrossRef]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; von Hohenberg, W.C.; Mickelson, D.J.; Lanier, L.M.; Georgieff, M.K. Iron Deficiency Impairs Developing Hippocampal Neuron Gene Expression, Energy Metabolism, and Dendrite Complexity. Dev. Neurosci. 2016, 38, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; von Hohenberg, W.C.; Georgieff, M.K.; Lanier, L.M. Chronic Energy Depletion due to Iron Deficiency Impairs Dendritic Mitochondrial Motility during Hippocampal Neuron Development. J. Neurosci. 2019, 39, 802–813. [Google Scholar] [CrossRef]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Simmons, E.C.; Scholpa, N.E.; Schnellmann, R.G. Mitochondrial biogenesis as a therapeutic target for traumatic and neurodegenerative CNS diseases. Exp. Neurol. 2020, 329, 113309. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, J.; Duarte, A.I. Overcoming mitochondrial dysfunction in neurodegenerative diseases. Neural Regen. Res. 2023, 18, 1486–1488. [Google Scholar] [CrossRef]
- Davis, G.W. Not Fade Away: Mechanisms of Neuronal ATP Homeostasis. Neuron 2020, 105, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Johnson-Cadwell, L.; Vesce, S.; Jekabsons, M.; Yadava, N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J. Neurosci. Res. 2007, 85, 3206–3212. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. Elife 2016, 5, e13378. [Google Scholar] [CrossRef]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348.e10. [Google Scholar] [CrossRef] [PubMed]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Painuli, S.; Semwal, P.; Zam, W.; Taheri, Y.; Ezzat, S.M.; Zuo, P.; Li, L.; Kumar, D.; Sharifi-Rad, J.; Cruz-Martins, N. NMDA Inhibitors: A Potential Contrivance to Assist in Management of Alzheimer’s Disease. Comb. Chem. High Throughput Screen. 2023, 26, 2099–2112. [Google Scholar] [CrossRef]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and myelination: The role of iron. Glia 2009, 57, 467–478. [Google Scholar] [CrossRef]
- Stephenson, E.; Nathoo, N.; Mahjoub, Y.; Dunn, J.F.; Yong, V.W. Iron in multiple sclerosis: Roles in neurodegeneration and repair. Nat. Rev. Neurol. 2014, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xie, Y.; Wang, X.; Abboud, M.I.; Ma, C.; Ge, W.; Schofield, C.J. Exploring links between 2-oxoglutarate-dependent oxygenases and Alzheimer’s disease. Alzheimer’s Dement. 2022, 18, 2637–2668. [Google Scholar] [CrossRef]
- LeVine, S.M.; Tsau, S.; Gunewardena, S. Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer’s Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration. Brain Sci. 2023, 13, 511. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Crooks, D.R.; Wilson-Ollivierre, H.; Ghosh, M.C.; Sougrat, R.; Lee, J.; Cooperman, S.; Mitchell, J.B.; Beaumont, C.; Rouault, T.A. Iron insufficiency compromises motor neurons and their mitochondrial function in Irp2-null mice. PLoS ONE 2011, 6, e25404. [Google Scholar] [CrossRef]
- Fretham, S.J.B.; Carlson, E.S.; Wobken, J.; Tran, P.V.; Petryk, A.; Georgieff, M.K. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus 2012, 22, 1691–1702. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef]
- Segatori, L. Impairment of homeostasis in lysosomal storage disorders. IUBMB Life 2014, 66, 472–477. [Google Scholar] [CrossRef]
- Pandey, M.K. Exploring Pro-Inflammatory Immunological Mediators: Unraveling the Mechanisms of Neuroinflammation in Lysosomal Storage Diseases. Biomedicines 2023, 11, 1067. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.L.; Tecedor, L.; Chang, M.; Davidson, B.L. Clarifying lysosomal storage diseases. Trends Neurosci. 2011, 34, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.M.; Ke, Y. Brain iron transport. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1672–1684. [Google Scholar] [CrossRef] [PubMed]
- Skjørringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Huang, Y.J.; Cui, J.T.; Song, N.; Xie, J. Iron Dysregulation in Parkinson’s Disease: Focused on the Autophagy–Lysosome Pathway. ACS Chem. Neurosci. 2019, 10, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife 2019, 8, e51031. [Google Scholar] [CrossRef] [PubMed]
- Rizzollo, F.; More, S.; Vangheluwe, P.; Agostinis, P. The lysosome as a master regulator of iron metabolism. Trends Biochem. Sci. 2021, 46, 960–975. [Google Scholar] [CrossRef]
- Clark, L.N.; Chan, R.; Cheng, R.; Liu, X.; Park, N.; Parmalee, N.; Kisselev, S.; Cortes, E.; Torres, P.A.; Pastores, G.M.; et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS ONE 2015, 10, e0125204. [Google Scholar] [CrossRef]
- Boudewyn, L.C.; Walkley, S.U. Current concepts in the neuropathogenesis of mucolipidosis type IV. J. Neurochem. 2019, 148, 669–689. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kosman, D.J. Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons. J. Neurochem. 2015, 133, 668–683. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Pasquadibisceglie, A.; Bonaccorsi di Patti, M.C.; Musci, G.; Polticelli, F. Membrane Transporters Involved in Iron Trafficking: Physiological and Pathological Aspects. Biomolecules 2023, 13, 1172. [Google Scholar] [CrossRef] [PubMed]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef]
- Allen, G.F.G.; Toth, R.; James, J.; Ganley, I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wei, H.; Zhang, D.; Sehgal, S.A.; Zhang, D.; Wang, X.; Qin, Y.; Liu, L.; Chen, Q. Defective mitochondrial ISCs biogenesis switches on IRP1 to fine tune selective mitophagy. Redox Biol. 2020, 36, 101661. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Paliakkara, J.J.; Melman, G.; Melman, A. Reductive Mobilization of Iron from Intact Ferritin: Mechanisms and Physiological Implication. Pharmaceuticals 2018, 11, 120. [Google Scholar] [CrossRef]
- Badu-Boateng, C.; Naftalin, R.J. Ascorbate and ferritin interactions: Consequences for iron release in vitro and in vivo and implications for inflammation. Free Radic. Biol. Med. 2019, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, S.; Scotto-Rosato, A.; Medina, D.L. TRPML1: The Ca(2+)retaker of the lysosome. Cell Calcium 2018, 69, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Knupp, A.; Mishra, S.; Martinez, R.; Braggin, J.E.; Szabo, M.; Kinoshita, C.; Hailey, D.W.; Small, S.A.; Jayadev, S.; Young, J.E. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020, 31, 107719. [Google Scholar] [CrossRef]
- Hung, C.; Tuck, E.; Stubbs, V.; van der Lee, S.J.; Aalfs, C.; van Spaendonk, R.; Scheltens, P.; Hardy, J.; Holstege, H.; Livesey, F.J. SORL1 deficiency in human excitatory neurons causes APP-dependent defects in the endolysosome-autophagy network. Cell Rep. 2021, 35, 109259. [Google Scholar] [CrossRef]
- Barthelson, K.; Pederson, S.M.; Newman, M.; Lardelli, M. Brain transcriptome analysis reveals subtle effects on mitochondrial function and iron homeostasis of mutations in the SORL1 gene implicated in early onset familial Alzheimer’s disease. Mol. Brain. 2020, 13, 142. [Google Scholar] [CrossRef]
- Mishra, S.; Knupp, A.; Szabo, M.P.; Williams, C.A.; Kinoshita, C.; Hailey, D.W.; Wang, Y.; Andersen, O.M.; Young, J.E. The Alzheimer’s gene SORL1 is a regulator of endosomal traffic and recycling in human neurons. Cell. Mol. Life Sci. 2022, 79, 162. [Google Scholar] [CrossRef]
- Misko, A.; Wood, L.; Kiselyov, K.; Slaugenhaupt, S.; Grishchuk, Y. Progress in elucidating pathophysiology of mucolipidosis IV. Neurosci. Lett. 2021, 755, 135944. [Google Scholar] [CrossRef]
- Zerem, A.; Ben-Sira, L.; Vigdorovich, N.; Leibovitz, Z.; Fisher, Y.; Schiffmann, R.; Grishchuk, Y.; Misko, A.L.; Orenstein, N.; Lev, D.; et al. White matter abnormalities and iron deposition in prenatal mucolipidosis IV—Fetal imaging and pathology. Metab. Brain Dis. 2021, 36, 2155–2167. [Google Scholar] [CrossRef]
- Frei, K.P.; Patronas, N.J.; Crutchfield, K.E.; Altarescu, G.; Schiffmann, R. Mucolipidosis type IV: Characteristic MRI findings. Neurology 1998, 51, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Dwyer, N.K.; Lubensky, I.A.; Tsokos, M.; Sutliff, V.E.; Latimer, J.S.; Frei, K.P.; Brady, R.O.; Barton, N.W.; Blanchette-Mackie, E.J.; et al. Constitutive achlorhydria in mucolipidosis type IV. Proc. Natl. Acad. Sci. USA 1998, 95, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Lubensky, I.A.; Schiffmann, R.; Goldin, E.; Tsokos, M. Lysosomal inclusions in gastric parietal cells in mucolipidosis type IV: A novel cause of achlorhydria and hypergastrinemia. Am. J. Surg. Pathol. 1999, 23, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, D.A.; Frumkin, A.; Bach, G. TRPML and lysosomal function. Biochim. Biophys. Acta 2007, 1772, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.P.; Wang, X.; Shen, D.; Chen, S.; Liu, M.; Wang, Y.; Mills, E.; Cheng, X.; Delling, M.; Xu, H. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J. Biol. Chem. 2009, 284, 32040–32052. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Raychowdhury, M.K.; Gonzalez-Perrett, S.; Montalbetti, N.; Timpanaro, G.A.; Chasan, B.; Goldmann, W.H.; Stahl, S.; Cooney, A.; Goldin, E.; Cantiello, H.F. Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. Hum. Mol. Genet. 2004, 13, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Bach, G.; Chen, C.S.; Pagano, R.E. Elevated lysosomal pH in Mucolipidosis type IV cells. Clin. Chim. Acta 1999, 280, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Soyombo, A.A.; Tjon-Kon-Sang, S.; Rbaibi, Y.; Bashllari, E.; Bisceglia, J.; Muallem, S.; Kiselyov, K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J. Biol. Chem. 2006, 281, 7294–7301. [Google Scholar] [CrossRef] [PubMed]
- Kogot-Levin, A.; Zeigler, M.; Ornoy, A.; Bach, G. Mucolipidosis type IV: The effect of increased lysosomal pH on the abnormal lysosomal storage. Pediatr. Res. 2009, 65, 686–690. [Google Scholar] [CrossRef]
- Folts, C.J.; Scott-Hewitt, N.; Pröschel, C.; Mayer-Pröschel, M.; Noble, M. Lysosomal Re-acidification Prevents Lysosphingolipid-Induced Lysosomal Impairment and Cellular Toxicity. PLoS Biol. 2016, 14, e1002583. [Google Scholar] [CrossRef] [PubMed]
- Kuk, M.U.; Lee, Y.H.; Kim, J.W.; Hwang, S.Y.; Park, J.T.; Park, S.C. Potential Treatment of Lysosomal Storage Disease through Modulation of the Mitochondrial—Lysosomal Axis. Cells 2021, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca2+ dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef] [PubMed]
- Coblentz, J.; St Croix, C.; Kiselyov, K. Loss of TRPML1 promotes production of reactive oxygen species: Is oxidative damage a factor in mucolipidosis type IV? Biochem. J. 2014, 457, 361–368. [Google Scholar] [CrossRef]
- Grishchuk, Y.; Peña, K.A.; Coblentz, J.; King, V.E.; Humphrey, D.M.; Wang, S.L.; Kiselyov, K.I.; Slaugenhaupt, S.A. Impaired myelination and reduced brain ferric iron in the mouse model of mucolipidosis IV. Dis. Model Mech. 2015, 8, 1591–1601. [Google Scholar] [CrossRef]
- Mepyans, M.; Andrzejczuk, L.; Sosa, J.; Smith, S.; Herron, S.; DeRosa, S.; Slaugenhaupt, S.A.; Misko, A.; Grishchuk, Y.; Kiselyov, K. Early evidence of delayed oligodendrocyte maturation in the mouse model of mucolipidosis type IV. Dis. Model Mech. 2020, 13, dmm044230. [Google Scholar] [CrossRef]
- LeVine, S.M.; Macklin, W.B. Iron-enriched oligodendrocytes: A reexamination of their spatial distribution. J. Neurosci. Res. 1990, 26, 508–512. [Google Scholar] [CrossRef]
- LeVine, S.M. Oligodendrocytes and myelin sheaths in normal, quaking and shiverer brains are enriched in iron. J. Neurosci. Res. 1991, 29, 413–419. [Google Scholar] [CrossRef]
- Oloyede, O.B.; Folayan, A.T.; Odutuga, A.A. Effects of low-iron status and deficiency of essential fatty acids on some biochemical constituents of rat brain. Biochem. Int. 1992, 27, 913–922. [Google Scholar]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- De Faria, O., Jr.; Pivonkova, H.; Varga, B.; Timmler, S.; Evans, K.A.; Káradóttir, R.T. Periods of synchronized myelin changes shape brain function and plasticity. Nat. Neurosci. 2021, 24, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Vanier, M.T. Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2015, 38, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Meske, V.; Erz, J.; Priesnitz, T.; Ohm, T.G. The autophagic defect in Niemann-Pick disease type C neurons differs from somatic cells and reduces neuronal viability. Neurobiol. Dis. 2014, 64, 88–97. [Google Scholar] [CrossRef]
- Liedtke, M.; Völkner, C.; Hermann, A.; Frech, M.J. Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann-Pick Disease Type C. Cells 2022, 11, 507. [Google Scholar] [CrossRef]
- Gondré-Lewis, M.C.; McGlynn, R.; Walkley, S.U. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr. Biol. 2003, 13, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Trilck, M.; Peter, F.; Zheng, C.; Frank, M.; Dobrenis, K.; Mascher, H.; Rolfs, A.; Frech, M.J. Diversity of glycosphingolipid GM2 and cholesterol accumulation in NPC1 patient-specific iPSC-derived neurons. Brain Res. 2017, 1657, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Williams, I.; Smith, D.; Cox, T.M.; Platt, F.M. Critical role of iron in the pathogenesis of the murine gangliosidoses. Neurobiol. Dis. 2009, 34, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Ravanfar, P.; Syeda, W.T.; Rushmore, R.J.; Moffat, B.; Lyall, A.E.; Merritt, A.H.; Devenyi, G.A.; Chakravarty, M.M.; Desmond, P.; Cropley, V.L.; et al. Investigation of Brain Iron in Niemann-Pick Type C: A 7T Quantitative Susceptibility Mapping Study. AJNR Am. J. Neuroradiol. 2023, 44, 768–775. [Google Scholar] [CrossRef]
- Hung, Y.H.; Faux, N.G.; Killilea, D.W.; Yanjanin, N.; Firnkes, S.; Volitakis, I.; Ganio, G.; Walterfang, M.; Hastings, C.; Porter, F.D.; et al. Altered transition metal homeostasis in Niemann-Pick disease, type C1. Metallomics 2014, 6, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.H.; Lotan, A.; Yeshurun, S.; Schroeder, A.; Bush, A.I. Iron chelation by deferiprone does not rescue the Niemann-Pick Disease Type C1 mouse model. Biometals 2020, 33, 87–95. [Google Scholar] [CrossRef]
- Martins, T.S.; Costa, R.S.; Vilaça, R.; Lemos, C.; Teixeira, V.; Pereira, C.; Costa, V. Iron Limitation Restores Autophagy and Increases Lifespan in the Yeast Model of Niemann-Pick Type C1. Int. J. Mol. Sci. 2023, 24, 6221. [Google Scholar] [CrossRef]
- Christomanou, H.; Vanier, M.T.; Santambrogio, P.; Arosio, P.; Kleijer, W.J.; Harzer, K. Deficient ferritin immunoreactivity in tissues from niemann-pick type C patients: Extension of findings to fetal tissues, H and L ferritin isoforms, but also one case of the rare Niemann-Pick C2 complementation group. Mol. Genet. Metab. 2000, 70, 196–202. [Google Scholar] [CrossRef]
- Liang, L.; Wang, H.; Yao, J.; Wei, Q.; Lu, Y.; Wang, T.; Cao, X. NPC1 Deficiency Contributes to Autophagy-Dependent Ferritinophagy in HEI-OC1 Auditory Cells. Front. Mol. Biosci. 2022, 9, 952608. [Google Scholar] [CrossRef]
- Iwai, K.; Drake, S.K.; Wehr, N.B.; Weissman, A.M.; LaVaute, T.; Minato, N.; Klausner, R.D.; Levine, R.L.; Rouault, T.A. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: Implications for degradation of oxidized proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 4924–4928. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef]
- Kidane, T.Z.; Sauble, E.; Linder, M.C. Release of iron from ferritin requires lysosomal activity. Am. J. Physiol. Cell Physiol. 2006, 291, C445–C455. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.C.W.; Siebel, S.; Colaco, A.; Nicoli, E.R.; Platt, N.; Shepherd, D.; Newman, S.; Armitage, A.E.; Farhat, N.Y.; Seligmann, G.; et al. Defective iron homeostasis and hematological abnormalities in Niemann-Pick disease type C1. Wellcome Open Res. 2023, 7, 267. [Google Scholar] [CrossRef] [PubMed]
- Cervera Bravo, A.; Osuna Marco, M.P.; Morán-Jiménez, M.J.; Martín-Hernández, E. Unexpected Cause of Persistent Microcytosis and Neurological Symptoms in a Child: Niemann-Pick Disease Type C. J. Pediatr. Hematol. Oncol. 2021, 43, e1238–e1240. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; De Nuccio, C.; Visentin, S.; Martire, A.; Minghetti, L.; Popoli, P.; Ferrante, A. Myelin Defects in Niemann-Pick Type C Disease: Mechanisms and Possible Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 8858. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.M.; Grubman, A.; Meyerowitz, J.; Duncan, C.; Tan, J.-L.; Parker, S.J.; Crouch, P.J.; Paterson, B.M.; Hickey, J.L.; Donnelly, P.S.; et al. Increased zinc and manganese in parallel with neurodegeneration, synaptic protein changes and activation of Akt/GSK3 signaling in ovine CLN6 neuronal ceroid lipofuscinosis. PLoS ONE 2013, 8, e58644. [Google Scholar] [CrossRef]
- Grubman, A.; Pollari, E.; Duncan, C.; Caragounis, A.; Blom, T.; Volitakis, I.; Wong, A.; Cooper, J.; Crouch, P.J.; Koistinaho, J.; et al. Deregulation of biometal homeostasis: The missing link for neuronal ceroid lipofuscinoses? Metallomics 2014, 6, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Guennel, P.; Vessey, K.A.; Jones, M.W.M.; James, S.A.; de Jonge, M.D.; White, A.R.; Fletcher, E.L. X-ray fluorescence microscopic measurement of elemental distribution in the mouse retina with age. Metallomics 2016, 8, 1110–1121. [Google Scholar] [CrossRef]
- Bagh, M.B.; Peng, S.; Chandra, G.; Zhang, Z.; Singh, S.P.; Pattabiraman, N.; Liu, A.; Mukherjee, A.B. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat. Commun. 2017, 8, 14612. [Google Scholar] [CrossRef]
- Naseri, N.; Sharma, M.; Velinov, M. Autosomal dominant neuronal ceroid lipofuscinosis: Clinical features and molecular basis. Clin. Genet. 2021, 99, 111–118. [Google Scholar] [CrossRef]
- Naseri, N.N.; Ergel, B.; Kharel, P.; Na, Y.; Huang, Q.; Huang, R.; Dolzhanskaya, N.; Burré, J.; Velinov, M.T.; Sharma, M. Aggregation of mutant cysteine string protein-α via Fe–S cluster binding is mitigated by iron chelators. Nat. Struct. Mol. Biol. 2020, 27, 192–201. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef]
- Tong, W.-H.; Rouault, T.A. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006, 3, 199–210. [Google Scholar] [CrossRef]
- Pastores, G.M.; Hughes, D.A. Gaucher Disease. In GeneReviews®; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Pelled, D.; Riebeling, C.; Bodennec, J.; de-Morgan, A.; Waller, H.; Schiffmann, R.; Futerman, A.H. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 2003, 278, 23594–23599. [Google Scholar] [CrossRef]
- Pelled, D.; Trajkovic-Bodennec, S.; Lloyd-Evans, E.; Sidransky, E.; Schiffmann, R.; Futerman, A.H. Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol. Dis. 2005, 18, 83–88. [Google Scholar] [CrossRef]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, M.P.; Jones, J.W.; Kane, M.; Awad, O.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1. Stem Cells Transl. Med. 2021, 10, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Braunstein, H.; Zimran, A.; Revel-Vilk, S.; Goker-Alpan, O. Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv. Drug Deliv. Rev. 2022, 187, 114402. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Subramanian, S.; Weinreb, N.J.; Kartha, R.V. Gaucher disease—More than just a rare lipid storage disease. J. Mol. Med. 2022, 100, 499–518. [Google Scholar] [CrossRef]
- Hruska, K.S.; Goker-Alpan, O.; Sidransky, E. Gaucher disease and the synucleinopathies. J. Biomed. Biotechnol. 2006, 2006, 78549. [Google Scholar] [CrossRef]
- Furderer, M.L.; Hertz, E.; Lopez, G.J.; Sidransky, E. Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism. Int. J. Mol. Sci. 2022, 23, 5842. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain-synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease. Mov. Disord. 2016, 31, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium inhibits spontaneous and iron-induced aggregation of α-synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef]
- Bharathi Rao, K.S. Thermodynamics imprinting reveals differential binding of metals to α-synuclein: Relevance to Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 359, 115–120. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, C.; Xu, H.H.; Liu, Y.N.; Zhou, F. Binding of α-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J. Inorg. Biochem. 2010, 104, 365–370. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Brandel, J.P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef]
- Castellani, R.J.; Siedlak, S.L.; Perry, G.; Smith, M.A. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000, 100, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and α-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 2019, 141, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef] [PubMed]
- Harvengt, J.; Wanty, C.; De Paepe, B.; Sempoux, C.; Revencu, N.; Smet, J.; Van Coster, R.; Lissens, W.; Seneca, S.; Weekers, L.; et al. Clinical variability in neurohepatic syndrome due to combined mitochondrial DNA depletion and Gaucher disease. Mol. Genet. Metab. Rep. 2014, 1, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Saffari, A.; Kölker, S.; Hoffmann, G.F.; Ebrahimi-Fakhari, D. Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 2017, 40, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.-C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef]
- Igisu, H.; Hamasaki, N.; Ito, A.; Ou, W. Inhibition of cytochrome c oxidase and hemolysis caused by lysosphingolipids. Lipids 1988, 23, 345–348. [Google Scholar] [CrossRef]
- De Deungria, M.; Rao, R.; Wobken, J.D.; Luciana, M.; Nelson, C.A.; Georgieff, M.K. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr. Res. 2000, 48, 169–176. [Google Scholar] [CrossRef]
- Rineau, E.; Gaillard, T.; Gueguen, N.; Procaccio, V.; Henrion, D.; Prunier, F.; Lasocki, S. Iron deficiency without anemia is responsible for decreased left ventricular function and reduced mitochondrial complex I activity in a mouse model. Int. J. Cardiol. 2018, 266, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Rineau, E.; Gueguen, N.; Procaccio, V.; Geneviève, F.; Reynier, P.; Henrion, D.; Lasocki, S. Iron Deficiency without Anemia Decreases Physical Endurance and Mitochondrial Complex I Activity of Oxidative Skeletal Muscle in the Mouse. Nutrients 2021, 13, 1056. [Google Scholar] [CrossRef]
- Chung, Y.J.; Swietach, P.; Curtis, M.K.; Ball, V.; Robbins, P.A.; Lakhal-Littleton, S. Iron-deficiency anemia results in transcriptional and metabolic remodeling in the heart toward a glycolytic phenotype. Front. Cardiovasc. Med. 2021, 7, 616920. [Google Scholar] [CrossRef]
- Stein, P.; Yu, H.; Jain, D.; Mistry, P.K. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am. J. Hematol. 2010, 85, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Stirnemann, J.; Belmatoug, N.; Heraoui, D.; Fantin, B.; Fain, O.; Charpentier, A.; Rose, C. Ferritinemia during type 1 Gaucher disease: Mechanisms and progression under treatment. Blood Cells Mol. Dis. 2012, 49, 53–57. [Google Scholar] [CrossRef]
- Lefebvre, T.; Reihani, N.; Daher, R.; de Villemeur, T.B.; Belmatoug, N.; Rose, C.; Colin-Aronovicz, Y.; Puy, H.; Le Van Kim, C.; Franco, M.; et al. Involvement of hepcidin in iron metabolism dysregulation in Gaucher disease. Haematologica 2018, 103, 587–596. [Google Scholar] [CrossRef]
- Lorenz, F.; Pawłowicz, E.; Klimkowska, M.; Beshara, S.; Bulanda Brustad, A.; Skotnicki, A.B.; Wahlin, A.; Machaczka, M. Ferritinemia and serum inflammatory cytokines in Swedish adults with Gaucher disease type 1. Blood Cells Mol. Dis. 2018, 68, 35–42. [Google Scholar] [CrossRef]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef]
- Domellöf, M.; Braegger, C.; Campoy, C.; Colomb, V.; Decsi, T.; Fewtrell, M.; Hojsak, I.; Mihatsch, W.; Molgaard, C.; Shamir, R.; et al. Iron requirements of infants and toddlers. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 119–129. [Google Scholar] [CrossRef]
- Beard, J. Iron deficiency alters brain development and functioning. J. Nutr. 2003, 133, 1468S–1472S. [Google Scholar] [CrossRef]
- Beard, J.L. Why iron deficiency is important in infant development. J. Nutr. 2008, 138, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, L.A.; Wobken, J.D.; Georgieff, M.K. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev. Neurosci. 2003, 25, 412–420. [Google Scholar] [CrossRef]
- Shah, H.E.; Bhawnani, N.; Ethirajulu, A.; Alkasabera, A.; Onyali, C.B.; Anim-Koranteng, C.; Mostafa, J.A. Iron Deficiency-Induced Changes in the Hippocampus, Corpus Striatum, and Monoamines Levels That Lead to Anxiety, Depression, Sleep Disorders, and Psychotic Disorders. Cureus 2021, 13, e18138. [Google Scholar] [CrossRef]
- Dallman, P.R.; Siimes, M.A.; Manies, E.C. Brain iron: Persistent deficiency following short-term iron deprivation in the young rat. Br. J. Haematol. 1975, 31, 209–215. [Google Scholar] [CrossRef]
- Erikson, K.M.; Pinero, D.J.; Connor, J.R.; Beard, J.L. Regional brain iron, ferritin and transferrin concentrations during iron deficiency and iron repletion in developing rats. J. Nutr. 1997, 127, 2030–2038. [Google Scholar] [CrossRef]
- Hu, X.; Wang, R.; Shan, Z.; Dong, Y.; Zheng, H.; Jesse, F.F.; Rao, E.; Takahashi, E.; Li, W.; Teng, W.; et al. Perinatal Iron Deficiency-Induced Hypothyroxinemia Impairs Early Brain Development Regardless of Normal Iron Levels in the Neonatal Brain. Thyroid 2016, 26, 891–900. [Google Scholar] [CrossRef]
- Barks, A.; Beeson, M.M.; Hallstrom, T.C.; Georgieff, M.K.; Tran, P.V. Developmental Iron Deficiency Dysregulates TET Activity and DNA Hydroxymethylation in the Rat Hippocampus and Cerebellum. Dev. Neurosci. 2022, 44, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Huff, T.C.; Wang, G. Epigenomic regulation by labile iron. Free Radic. Biol. Med. 2021, 170, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Lien, Y.-C.; Condon, D.E.; Georgieff, M.K.; Simmons, R.A.; Tran, P.V. Dysregulation of Neuronal Genes by Fetal-Neonatal Iron Deficiency Anemia Is Associated with Altered DNA Methylation in the Rat Hippocampus. Nutrients 2019, 11, 1191. [Google Scholar] [CrossRef]
- de Mello, A.S.; da Silva, I.R.V.; Reinaldo, G.P.; Dorneles, G.P.; Cé, J.; Lago, P.D.; Peres, A.; Elsner, V.R.; Coelho, J.C. The modulation of inflammatory parameters, Brain-derived neurotrophic factor levels and global histone H4 acetylation status in peripheral blood of patients with Gaucher disease type 1. Clin. Biochem. 2017, 50, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Wesnes, K.A.; Luzy, C.; Petakov, M.; Mrsic, M.; Niederau, C.; Giraldo, P.; Hughes, D.; Mehta, A.; Mengel, K.E.; et al. The cognitive profile of type 1 Gaucher disease patients. J. Inherit. Metab. Dis. 2012, 35, 1093–1099. [Google Scholar] [CrossRef]
- Tullo, M.G.; Cerulli Irelli, E.; Caramia, F.; Tessari, G.; Di Bonaventura, C.; Turchetta, R.; Giallonardo, A.T.; Palumbo, G.; Bianchi, S.; Atturo, F.; et al. The Spectrum of Neurological and Sensory Abnormalities in Gaucher Disease Patients: A Multidisciplinary Study (SENOPRO). Int. J. Mol. Sci. 2023, 24, 8844. [Google Scholar] [CrossRef]
- Liu, S.X.; Barks, A.K.; Lunos, S.; Gewirtz, J.C.; Georgieff, M.K.; Tran, P.V. Prenatal Iron Deficiency and Choline Supplementation Interact to Epigenetically Regulate Jarid1b and Bdnf in the Rat Hippocampus into Adulthood. Nutrients 2021, 13, 4527. [Google Scholar] [CrossRef]
- Lee, G.H.; D’Arcangelo, G. New Insights into Reelin-Mediated Signaling Pathways. Front. Cell. Neurosci. 2016, 10, 122. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y. Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules 2020, 10, 964. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Twenty five years of the “psychosine hypothesis”: A personal perspective of its history and present status. Neurochem. Res. 1998, 23, 251–259. [Google Scholar] [CrossRef]
- Voccoli, V.; Tonazzini, I.; Signore, G.; Caleo, M.; Cecchini, M. Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis. 2014, 5, e1529. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Suzuki, K. The twitcher mouse: A model for Krabbe disease and for experimental therapies. Brain Pathol. 1995, 5, 249–258. [Google Scholar] [CrossRef]
- Smith, B.R.; Santos, M.B.; Marshall, M.S.; Cantuti-Castelvetri, L.; Lopez-Rosas, A.; Li, G.; van Breemen, R.; Claycomb, K.I.; Gallea, J.I.; Celej, M.S.; et al. Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 2014, 232, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Hatton, C.; Ghanem, S.S.; Koss, D.J.; Abdi, I.Y.; Gibbons, E.; Guerreiro, R.; Bras, J.; Kun-Rodrigues, C.; Singleton, A.; Hernandez, D.; et al. Prion-like α-synuclein pathology in the brain of infants with Krabbe disease. Brain 2022, 145, 1257–1263. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G. A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Neumann, D.P.; Goodall, G.J.; Gregory, P.A. The Quaking RNA-binding proteins as regulators of cell differentiation. Wiley Interdiscip. Rev. RNA 2022, 13, e1724. [Google Scholar] [CrossRef]
- LeVine, S.M.; Torres, M.V. Morphological features of degenerating oligodendrocytes in twitcher mice. Brain Res. 1992, 587, 348–352. [Google Scholar] [CrossRef]
- Sturrock, R.R. Myelination of the mouse corpus callosum. Neuropathol. Appl. Neurobiol. 1980, 6, 415–420. [Google Scholar] [CrossRef]
- Lin, D.-S.; Ho, C.-S.; Huang, Y.-W.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Wang, T.-J.; Yang, S.-J.; Chiang, M.-F. Impairment of Proteasome and Autophagy Underlying the Pathogenesis of Leukodystrophy. Cells 2020, 9, 1124. [Google Scholar] [CrossRef]
- Vantaggiato, L.; Shaba, E.; Carleo, A.; Bezzini, D.; Pannuzzo, G.; Luddi, A.; Piomboni, P.; Bini, L.; Bianchi, L. Neurodegenerative Disorder Risk in Krabbe Disease Carriers. Int. J. Mol. Sci. 2022, 23, 13537. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Markus, M.; Seetulsingh, S.P.; Wrigglesworth, J.M. Kinetics of inhibition of purified and mitochondrial cytochrome c oxidase by psychosine (β-galactosylsphingosine). Biochem. J. 1993, 290, 139–144. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification—The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing. Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Prasad, H.; Rao, R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. USA 2018, 115, E6640–E6649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J. Neurosci. 2019, 39, 5255–5268. [Google Scholar] [CrossRef]
- Delport, A.; Hewer, R. The amyloid precursor protein: A converging point in Alzheimer’s disease. Mol. Neurobiol. 2022, 59, 4501–4516. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Rogers, J.T.; Majd, S.; Newman, M.; Sutherland, G.T.; Verdile, G.; Lardelli, M. Dysregulation of Neuronal Iron Homeostasis as an Alternative Unifying Effect of Mutations Causing Familial Alzheimer’s Disease. Front. Neurosci. 2018, 12, 533. [Google Scholar] [CrossRef]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-lysosomal and autophagic dysfunction: A driving factor in Alzheimer’s disease? J. Neurochem. 2017, 140, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, M.; Chang, W. Iron dyshomeostasis and ferroptosis in Alzheimer’s disease: Molecular mechanisms of cell death and novel therapeutic drugs and targets for AD. Front. Pharmacol. 2022, 13, 983623. [Google Scholar] [CrossRef]
- Pergande, M.R.; Cougnoux, A.; Rathnayake, R.A.C.; Porter, F.D.; Cologna, S.M. Differential Proteomics Reveals miR-155 as a Novel Indicator of Liver and Spleen Pathology in the Symptomatic Niemann-Pick Disease, Type C1 Mouse Model. Molecules 2019, 24, 994. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Frey, W.H., 2nd. A role for heme in Alzheimer’s disease: Heme binds amyloid beta and has altered metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 11153–11158. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.E.; Stone, M.L.; Zhu, X.; Perry, G.; Smith, M.A. Heme deficiency in Alzheimer’s disease: A possible connection to porphyria. J. Biomed. Biotechnol. 2006, 2006, 24038. [Google Scholar] [CrossRef]
- Boopathi, S.; Kolandaivel, P. Fe2+ binding on amyloid β-peptide promotes aggregation. Proteins 2016, 84, 1257–1274. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Adlard, P.A. Untangling Tau and Iron: Exploring the Interaction Between Iron and Tau in Neurodegeneration. Front. Mol. Neurosci. 2018, 11, 276. [Google Scholar] [CrossRef]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.S.; Goldwurm, S.; Frisén, J.; Deierborg, T.; Roybon, L. Alpha-Synuclein Expression in the Oligodendrocyte Lineage: An In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell Rep. 2015, 5, 174–184. [Google Scholar] [CrossRef]
- Sharma, M.; Burré, J. α-Synuclein in synaptic function and dysfunction. Trends Neurosci. 2023, 46, 153–166. [Google Scholar] [CrossRef]
- Qiang, Y.X.; Deng, Y.T.; Zhang, Y.R.; Wang, H.F.; Zhang, W.; Dong, Q.; Feng, J.F.; Cheng, W.; Yu, J.T. Associations of blood cell indices and anemia with risk of incident dementia: A prospective cohort study of 313,448 participants. Alzheimer’s Dement. 2023, 19, 3965–3976. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; Rao, R.; Tran, P.V.; Georgieff, M.K. The Effects of Early-Life Iron Deficiency on Brain Energy Metabolism. Neurosci. Insights 2020, 15, 2633105520935104. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, Z.; Cui, A.; Zhu, Y.; Zhang, L.; Liu, J.; Shi, S.; Fu, C.; Han, X.; Gao, W.; et al. Increased Iron Deposition on Brain Quantitative Susceptibility Mapping Correlates with Decreased Cognitive Function in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1849–1857. [Google Scholar] [CrossRef]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Damulina, A.; Pirpamer, L.; Soellradl, M.; Sackl, M.; Tinauer, C.; Hofer, E.; Enzinger, C.; Gesierich, B.; Duering, M.; Ropele, S.; et al. Cross-sectional and Longitudinal Assessment of Brain Iron Level in Alzheimer Disease Using 3-T MRI. Radiology 2020, 296, 619–626. [Google Scholar] [CrossRef]
- Yang, A.; Du, L.; Gao, W.; Liu, B.; Chen, Y.; Wang, Y.; Liu, X.; Lv, K.; Zhang, W.; Xia, H.; et al. Associations of cortical iron accumulation with cognition and cerebral atrophy in Alzheimer’s disease. Quant. Imaging Med. Surg. 2022, 12, 4570–4586. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Shahid, S.; Hone-Blanchet, A.; Huang, S.; Wu, J.; Bisht, A.; Loring, D.; Goldstein, F.; Levey, A.; Crosson, B.; et al. Magnetic resonance evidence of increased iron content in subcortical brain regions in asymptomatic Alzheimer’s disease. Hum. Brain Mapp. 2023, 44, 3072–3083. [Google Scholar] [CrossRef]
- Georgieff, M.K. The role of iron in neurodevelopment: Fetal iron deficiency and the developing hippocampus. Biochem. Soc. Trans. 2008, 36, 1267–1271. [Google Scholar] [CrossRef]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seiça, R.M.; Moreira, P.I. Alzheimer’s Disease: From Mitochondrial Perturbations to Mitochondrial Medicine. Brain Pathol. 2016, 26, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.V.; Fretham, S.J.B.; Carlson, E.S.; Georgieff, M.K. Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr. Res. 2009, 65, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Brunette, K.E.; Tran, P.V.; Wobken, J.D.; Carlson, E.S.; Georgieff, M.K. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev. Neurosci. 2010, 32, 238–248. [Google Scholar] [CrossRef]
- Nishikura, N.; Hino, K.; Kimura, T.; Uchimura, Y.; Hino, S.; Nakao, M.; Maruo, Y.; Udagawa, J. Postweaning Iron Deficiency in Male Rats Leads to Long-Term Hyperactivity and Decreased Reelin Gene Expression in the Nucleus Accumbens. J. Nutr. 2020, 150, 212–221. [Google Scholar] [CrossRef]
- Barks, A.K.; Liu, S.X.; Georgieff, M.K.; Hallstrom, T.C.; Tran, P.V. Early-Life Iron Deficiency Anemia Programs the Hippocampal Epigenomic Landscape. Nutrients 2021, 13, 3857. [Google Scholar] [CrossRef]
- Cisneros-Franco, J.M.; Voss, P.; Thomas, M.E.; de Villers-Sidani, E. Critical periods of brain development. Handb. Clin. Neurol. 2020, 173, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Humeres, A.; Elgueta, C.; Kirkwood, A.; Hidalgo, C.; Núñez, M.T. Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 2011, 286, 13382–13392. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Humeres, A. Iron deficiency on neuronal function. Biometals 2012, 25, 825–835. [Google Scholar] [CrossRef]
- Walkley, S.U. Neurobiology and cellular pathogenesis of glycolipid storage diseases. Philos. Trans. R. Soc. B 2003, 358, 893–904. [Google Scholar] [CrossRef]
- Caeyenberghs, K.; Balschun, D.; Roces, D.P.; Schwake, M.; Saftig, P.; D’Hooge, R. Multivariate neurocognitive and emotional profile of a mannosidosis murine model for therapy assessment. Neurobiol. Dis. 2006, 23, 422–432. [Google Scholar] [CrossRef]
- Faldini, E.; Stroobants, S.; Lüllmann-Rauch, R.; Eckhardt, M.; Gieselmann, V.; Balschun, D.; D’Hooge, R. Telencephalic histopathology and changes in behavioural and neural plasticity in a murine model for metachromatic leukodystrophy. Behav. Brain Res. 2011, 222, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef]
- Cuestas Torres, D.M.; Cardenas, F.P. Synaptic plasticity in Alzheimer’s disease and healthy aging. Rev. Neurosci. 2020, 31, 245–268. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Spampinato, D. Alzheimer disease and neuroplasticity. Handb. Clin. Neurol. 2022, 184, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Khattar, N.; Triebswetter, C.; Kiely, M.; Ferrucci, L.; Resnick, S.M.; Spencer, R.G.; Bouhrara, M. Investigation of the association between cerebral iron content and myelin content in normative aging using quantitative magnetic resonance neuroimaging. Neuroimage 2021, 239, 118267. [Google Scholar] [CrossRef]
- McGahan, M.C.; Harned, J.; Mukunnemkeril, M.; Goralska, M.; Fleisher, L.; Ferrell, J.B. Iron alters glutamate secretion by regulating cytosolic aconitase activity. Am. J. Physiol. Cell Physiol. 2005, 288, C1117–C1124. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links with Alzheimer’s Disease. Front. Neurosci. 2020, 13, 1443. [Google Scholar] [CrossRef]
- Chavoshinezhad, S.; Beirami, E.; Izadpanah, E.; Feligioni, M.; Hassanzadeh, K. Molecular mechanism and potential therapeutic targets of necroptosis and ferroptosis in Alzheimer’s disease. Biomed. Pharmacother. 2023, 168, 115656. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.-H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Gangwar, P.; Yadav, A.; Yadav, B.; Rao, R.; Kaur, S.; Kumar, P.; Dhiman, M.; Taglialatela, G.; Mantha, A.K. Understanding the neuronal synapse and challenges associated with the mitochondrial dysfunction in mild cognitive impairment and Alzheimer’s disease. Mitochondrion 2023, 73, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; di Ronza, A.; Lotfi, P.; Sardiello, M. Lysosomes and Brain Health. Annu. Rev. Neurosci. 2018, 41, 255–276. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeVine, S.M. Examining the Role of a Functional Deficiency of Iron in Lysosomal Storage Disorders with Translational Relevance to Alzheimer’s Disease. Cells 2023, 12, 2641. https://doi.org/10.3390/cells12222641
LeVine SM. Examining the Role of a Functional Deficiency of Iron in Lysosomal Storage Disorders with Translational Relevance to Alzheimer’s Disease. Cells. 2023; 12(22):2641. https://doi.org/10.3390/cells12222641
Chicago/Turabian StyleLeVine, Steven M. 2023. "Examining the Role of a Functional Deficiency of Iron in Lysosomal Storage Disorders with Translational Relevance to Alzheimer’s Disease" Cells 12, no. 22: 2641. https://doi.org/10.3390/cells12222641
APA StyleLeVine, S. M. (2023). Examining the Role of a Functional Deficiency of Iron in Lysosomal Storage Disorders with Translational Relevance to Alzheimer’s Disease. Cells, 12(22), 2641. https://doi.org/10.3390/cells12222641