
A New Era of Integration between Multiomics and Spatio-Temporal Analysis for the Translation of EMT towards Clinical Applications in Cancer




Abstract
:
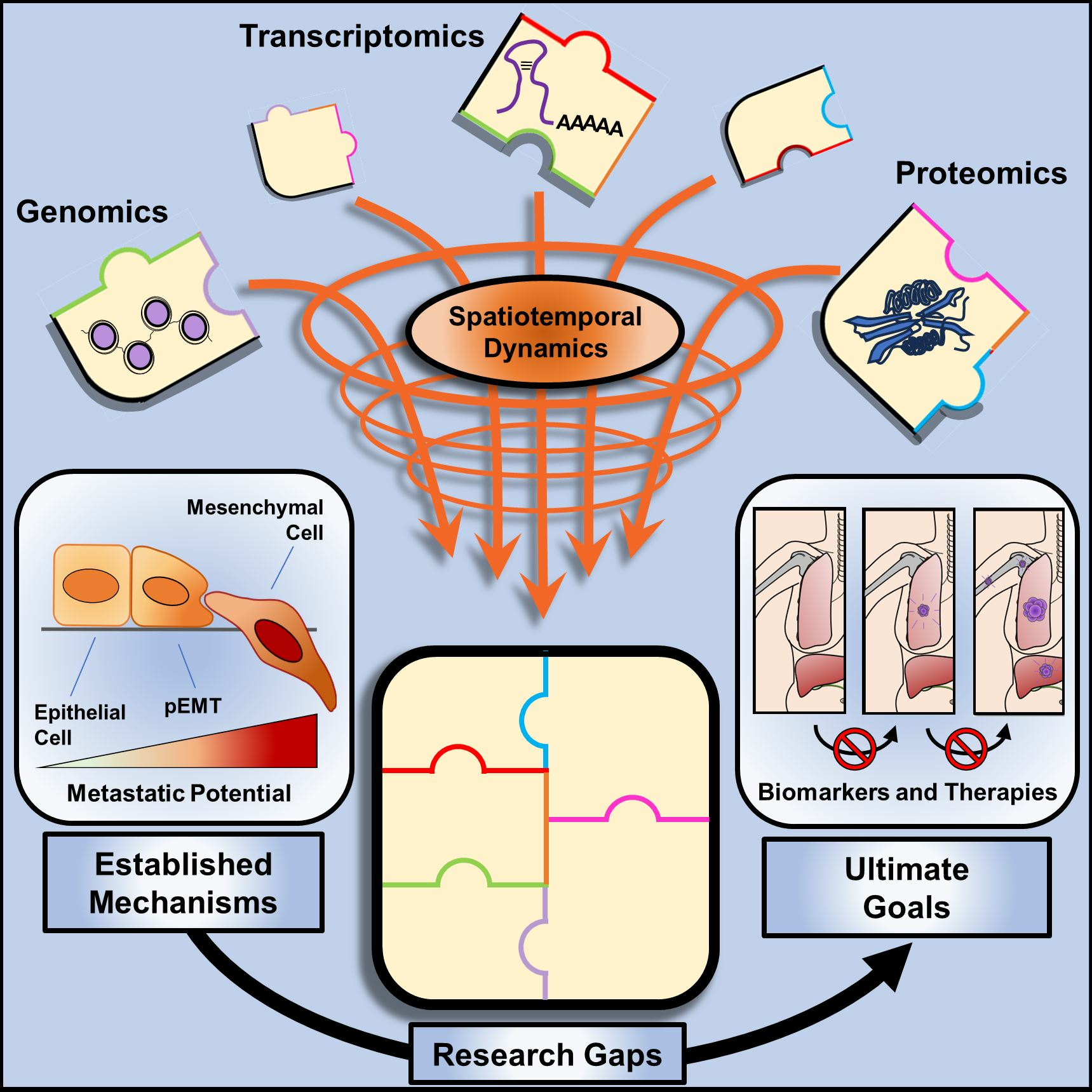{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction to EMT
1.1. Molecular Regulation of EMT
1.2. EMT as an ‘Accomplice’ to Cancer Metastasis
2. Benefits and Pitfalls of Analyzing Cancer Cell EMT at the DNA/RNA Level
3. Proteomics Translated from Bench-to-Bedside
3.1. Using In Vitro Models to Analyze the Proteome of Cancer Cells Undergoing EMT
3.1.1. Compartmentalization and Specificity of Sub-Proteome
3.1.2. What Is on the Outside Matters: Secretome and Cell Communication
3.2. Looking towards New EMT Biomarkers in Primary Tumors by Using Proteomics
3.3. Biological Fluids: An Easier Access to EMT-Related Biomarkers
4. Integration of Multiomics and Spatio-Temporal Analyses for a Comprehensive Understanding of EMT-Driven Cancer Progression
5. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Akhmetkaliyev, A.; Alibrahim, N.; Shafiee, D.; Tulchinsky, E. EMT/MET plasticity in cancer and Go-or-Grow decisions in quiescence: The two sides of the same coin? Mol. Cancer 2023, 22, 90. [Google Scholar] [CrossRef]
- Buckley, C.E.; St Johnston, D. Apical–basal polarity and the control of epithelial form and function. Nat. Rev. Mol. Cell Biol. 2022, 23, 559–577. [Google Scholar] [CrossRef]
- Lu, C.; Sidoli, S.; Kulej, K.; Ross, K.; Wu, C.H.; Garcia, B.A. Coordination between TGF-β cellular signaling and epigenetic regulation during epithelial to mesenchymal transition. Epigenetics Chromatin 2019, 12, 11. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e1624. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.P.; Sambath, J.; Sathe, G.; George, I.A.; Pandey, A.; Thompson, E.W.; Kumar, P. Pan-cancer quantitation of epithelial-mesenchymal transition dynamics using parallel reaction monitoring-based targeted proteomics approach. J. Transl. Med. 2022, 20, 84. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, S.; Adams, J.; De Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs 2021, 211, 157–182. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hong, W.; Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ng, A.S.; Cai, S.; Li, Q.; Yang, L.; Kerr, D. Novel therapeutic strategies: Targeting epithelial–mesenchymal transition in colorectal cancer. Lancet Oncol. 2021, 22, e358–e368. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef]
- Nam, M.W.; Kim, C.W.; Choi, K.C. Epithelial-Mesenchymal Transition-Inducing Factors Involved in the Progression of Lung Cancers. Biomol. Ther. 2022, 30, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Szymczyk, J.; Sluzalska, K.D.; Materla, I.; Opalinski, L.; Otlewski, J.; Zakrzewska, M. FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers 2021, 13, 5796. [Google Scholar] [CrossRef]
- Wu, S.; Luwor, R.B.; Zhu, H.-J. Dynamics of transforming growth factor β signaling and therapeutic efficacy. Growth Factors 2023, 41, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Vallés, A.M.; Boyer, B.; Badet, J.; Tucker, G.C.; Barritault, D.; Thiery, J.P. Acidic fibroblast growth factor is a modulator of epithelial plasticity in a rat bladder carcinoma cell line. Proc. Natl. Acad. Sci. USA 1990, 87, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef]
- Fan, C.; Wang, Q.; van der Zon, G.; Ren, J.; Agaser, C.; Slieker, R.C.; Iyengar, P.V.; Mei, H.; ten Dijke, P. OVOL1 inhibits breast cancer cell invasion by enhancing the degradation of TGF-β type I receptor. Signal Transduct. Target. Ther. 2022, 7, 126. [Google Scholar] [CrossRef]
- Lioulia, E.; Mokos, P.; Panteris, E.; Dafou, D. UBE2T promotes β-catenin nuclear translocation in hepatocellular carcinoma through MAPK/ERK-dependent activation. Mol. Oncol. 2022, 16, 1694–1713. [Google Scholar] [CrossRef]
- Mi, K.; Zeng, L.; Chen, Y.; Ning, J.; Zhang, S.; Zhao, P.; Yang, S. DHX38 enhances proliferation, metastasis, and EMT progression in NSCLC through the G3BP1-mediated MAPK pathway. Cell. Signal. 2024, 113, 110962. [Google Scholar] [CrossRef]
- Ma, X.; Ma, X.; Zhu, L.; Zhao, Y.; Chen, M.; Li, T.; Lin, Y.; Ma, D.; Sun, C.; Han, L. The E3 ubiquitin ligase MG53 inhibits hepatocellular carcinoma by targeting RAC1 signaling. Oncogenesis 2022, 11, 40. [Google Scholar] [CrossRef]
- Nie, X.; Liu, D.; Zheng, M.; Li, X.; Liu, O.; Guo, Q.; Zhu, L.; Lin, B. HERPUD1 promotes ovarian cancer cell survival by sustaining autophagy and inhibit apoptosis via PI3K/AKT/mTOR and p38 MAPK signaling pathways. BMC Cancer 2022, 22, 1338. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Zheng, H.; Kang, Y. Multilayer control of the EMT master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yun, F.; Shi, L.; Li, Z.H.; Luo, N.R.; Jia, Y.F. Roles of Signaling Pathways in the Epithelial-Mesenchymal Transition in Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 6201–6206. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-β signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.J.; Iaria, J.; Sizeland, A.M. Smad7 differentially regulates transforming growth factor β-mediated signaling pathways. J. Biol. Chem. 1999, 274, 32258–32264. [Google Scholar] [CrossRef] [PubMed]
- Khatibi, S.; Zhu, H.-J.; Wagner, J.; Tan, C.W.; Manton, J.H.; Burgess, A.W. Mathematical model of TGF-β signalling: Feedback coupling is consistent with signal switching. BMC Syst. Biol. 2017, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, M.; Hervouet, E.; Baudu, T.; Herfs, M.; Parratte, C.; Feugeas, J.P.; Perez, V.; Reynders, C.; Ancion, M.; Vigneron, M.; et al. GABARAPL1 Inhibits EMT Signaling through SMAD-Tageted Negative Feedback. Biology 2021, 10, 956. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal–epithelial transition in development and reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.N.; Jayachandran, G.; Moore, R.G.; Cristofanilli, M.; Lang, J.E.; Khoury, J.D.; Press, M.F.; Kim, K.K.; Khazan, N.; Zhang, Q.; et al. A Multi-Center Clinical Study to Harvest and Characterize Circulating Tumor Cells from Patients with Metastatic Breast Cancer Using the Parsortix® PC1 System. Cancers 2022, 14, 5238. [Google Scholar] [CrossRef] [PubMed]
- Boya, M.; Ozkaya-Ahmadov, T.; Swain, B.E.; Chu, C.-H.; Asmare, N.; Civelekoglu, O.; Liu, R.; Lee, D.; Tobia, S.; Biliya, S.; et al. High throughput, label-free isolation of circulating tumor cell clusters in meshed microwells. Nat. Commun. 2022, 13, 3385. [Google Scholar] [CrossRef]
- Gopal, S.K.; Greening, D.W.; Mathias, R.A.; Ji, H.; Rai, A.; Chen, M.; Zhu, H.J.; Simpson, R.J. YBX1/YB-1 induces partial EMT and tumourigenicity through secretion of angiogenic factors into the extracellular microenvironment. Oncotarget 2015, 6, 13718–13730. [Google Scholar] [CrossRef] [PubMed]
- Haerinck, J.; Berx, G. Partial EMT takes the lead in cancer metastasis. Dev. Cell 2021, 56, 3174–3176. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Abdollahi, B.; Wilkins, O.M.; Lu, H.; Chakraborty, P.; Ognjenovic, N.B.; Muller, K.E.; Jolly, M.K.; Christensen, B.C.; Hassanpour, S.; et al. Phenotypic heterogeneity driven by plasticity of the intermediate EMT state governs disease progression and metastasis in breast cancer. Sci. Adv. 2022, 8, eabj8002. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Wang, Q.; Kuipers, T.B.; Cats, D.; Iyengar, P.V.; Hagenaars, S.C.; Mesker, W.E.; Devilee, P.; Tollenaar, R.A.E.M.; Mei, H.; et al. LncRNA LITATS1 suppresses TGF-β-induced EMT and cancer cell plasticity by potentiating TβRI degradation. EMBO J. 2023, 42, e112806. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221.e3211. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e114. [Google Scholar] [CrossRef] [PubMed]
- Krol, I.; Schwab, F.D.; Carbone, R.; Ritter, M.; Picocci, S.; De Marni, M.L.; Stepien, G.; Franchi, G.M.; Zanardi, A.; Rissoglio, M.D.; et al. Detection of clustered circulating tumour cells in early breast cancer. Br. J. Cancer 2021, 125, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Koppaka, D.; Anand, A.; Deb, B.; Grenci, G.; Viasnoff, V.; Thompson, E.W.; Gowda, H.; Bhat, R.; Rangarajan, A.; et al. Circulating Tumor Cell cluster phenotype allows monitoring response to treatment and predicts survival. Sci. Rep. 2019, 9, 7933. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Storey, B.D.; Moore, J.C.; Tang, Q.; Chen, Y.-L.; Javaid, S.; Sarioglu, A.F.; Sullivan, R.; Madden, M.W.; O’Keefe, R.; et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. USA 2016, 113, 4947–4952. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-D.; Pang, K.; Wu, Z.-X.; Dong, Y.; Hao, L.; Qin, J.-X.; Wang, W.; Chen, Z.-S.; Han, C.-H. Tumor cell plasticity in targeted therapy-induced resistance: Mechanisms and new strategies. Signal Transduct. Target. Ther. 2023, 8, 113. [Google Scholar] [CrossRef]
- Kralj, J.; Pernar Kovač, M.; Dabelić, S.; Polančec, D.S.; Wachtmeister, T.; Köhrer, K.; Brozovic, A. Transcriptome analysis of newly established carboplatin-resistant ovarian cancer cell model reveals genes shared by drug resistance and drug-induced EMT. Br. J. Cancer 2023, 128, 1344–1359. [Google Scholar] [CrossRef] [PubMed]
- Billottet, C.; Tuefferd, M.; Gentien, D.; Rapinat, A.; Thiery, J.P.; Broet, P.; Jouanneau, J. Modulation of several waves of gene expression during FGF-1 induced epithelial-mesenchymal transition of carcinoma cells. J. Cell Biochem. 2008, 104, 826–839. [Google Scholar] [CrossRef]
- Lenferink, A.E.; Cantin, C.; Nantel, A.; Wang, E.; Durocher, Y.; Banville, M.; Paul-Roc, B.; Marcil, A.; Wilson, M.R.; O’Connor-McCourt, M.D. Transcriptome profiling of a TGF-β-induced epithelial-to-mesenchymal transition reveals extracellular clusterin as a target for therapeutic antibodies. Oncogene 2010, 29, 831–844. [Google Scholar] [CrossRef]
- Lu, Z.X.; Huang, Q.; Park, J.W.; Shen, S.; Lin, L.; Tokheim, C.J.; Henry, M.D.; Xing, Y. Transcriptome-wide landscape of pre-mRNA alternative splicing associated with metastatic colonization. Mol. Cancer Res. 2015, 13, 305–318. [Google Scholar] [CrossRef]
- Loboda, A.; Nebozhyn, M.V.; Watters, J.W.; Buser, C.A.; Shaw, P.M.; Huang, P.S.; Van’t Veer, L.; Tollenaar, R.A.E.M.; Jackson, D.B.; Agrawal, D.; et al. EMT is the dominant program in human colon cancer. BMC Med. Genom. 2011, 4, 9. [Google Scholar] [CrossRef]
- Baniwal, S.K.; Khalid, O.; Gabet, Y.; Shah, R.R.; Purcell, D.J.; Mav, D.; Kohn-Gabet, A.E.; Shi, Y.; Coetzee, G.A.; Frenkel, B. Runx2 transcriptome of prostate cancer cells: Insights into invasiveness and bone metastasis. Mol. Cancer 2010, 9, 258. [Google Scholar] [CrossRef]
- Frey, P.; Devisme, A.; Rose, K.; Schrempp, M.; Freihen, V.; Andrieux, G.; Boerries, M.; Hecht, A. SMAD4 mutations do not preclude epithelial–mesenchymal transition in colorectal cancer. Oncogene 2022, 41, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Malouf, G.G.; Lu, E.; Sphyris, N.; Vijay, V.; Ramachandran, P.P.; Ueno, K.R.; Gaur, S.; Nicoloso, M.S.; Rossi, S.; et al. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci. Rep. 2013, 3, 2687. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.E.; Ku, J.; Honda, T.K.; Yu, V.; Kuo, S.Z.; Zheng, H.; Xuan, Y.; Saad, M.A.; Hinton, A.; Brumund, K.T.; et al. Transcriptome sequencing uncovers novel long noncoding and small nucleolar RNAs dysregulated in head and neck squamous cell carcinoma. RNA 2015, 21, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Y.; Wu, J.; Wang, Y.J.; He, J.H.; Deng, W.X.; Hu, K.; Zhang, Y.C.; Zhang, Y.; Yan, H.; Wang, D.L.; et al. Deep sequencing reveals a global reprogramming of lncRNA transcriptome during EMT. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Seo, D.; Andersen, J.B.; Gillen, M.C.; Kim, M.S.; Conner, E.A.; Galle, P.R.; Factor, V.M.; Park, Y.N.; Thorgeirsson, S.S. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J. Hepatol. 2014, 60, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Szabo, P.M.; Vajdi, A.; Kumar, N.; Tolstorukov, M.Y.; Chen, B.J.; Edwards, R.; Ligon, K.L.; Chasalow, S.D.; Chow, K.H.; Shetty, A.; et al. Cancer-associated fibroblasts are the main contributors to epithelial-to-mesenchymal signatures in the tumor microenvironment. Sci. Rep. 2023, 13, 3051. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Tabuchi, Y.; Yurino, H.; Hirohashi, Y.; Deshimaru, S.; Asano, T.; Mariya, T.; Oshima, K.; Takamura, Y.; Ukita, Y.; et al. Comprehensive single-cell transcriptome analysis reveals heterogeneity in endometrioid adenocarcinoma tissues. Sci. Rep. 2017, 7, 14225. [Google Scholar] [CrossRef]
- Bocci, F.; Zhou, P.; Nie, Q. Single-Cell RNA-Seq Analysis Reveals the Acquisition of Cancer Stem Cell Traits and Increase of Cell–Cell Signaling during EMT Progression. Cancers 2021, 13, 5726. [Google Scholar] [CrossRef]
- Tyler, M.; Tirosh, I. Decoupling epithelial-mesenchymal transitions from stromal profiles by integrative expression analysis. Nat. Commun. 2021, 12, 2592. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef]
- Sutton, G.J.; Poppe, D.; Simmons, R.K.; Walsh, K.; Nawaz, U.; Lister, R.; Gagnon-Bartsch, J.A.; Voineagu, I. Comprehensive evaluation of deconvolution methods for human brain gene expression. Nat. Commun. 2022, 13, 1358. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Keshamouni, V.G.; Jagtap, P.; Michailidis, G.; Strahler, J.R.; Kuick, R.; Reka, A.K.; Papoulias, P.; Krishnapuram, R.; Srirangam, A.; Standiford, T.J.; et al. Temporal quantitative proteomics by iTRAQ 2D-LC-MS/MS and corresponding mRNA expression analysis identify post-transcriptional modulation of actin-cytoskeleton regulators during TGF-β-Induced epithelial-mesenchymal transition. J. Proteome Res. 2009, 8, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Ruff, M.; Leyme, A.; Le Cann, F.; Bonnier, D.; Le Seyec, J.; Chesnel, F.; Fattet, L.; Rimokh, R.; Baffet, G.; Theret, N. The Disintegrin and Metalloprotease ADAM12 Is Associated with TGF-β-Induced Epithelial to Mesenchymal Transition. PLoS ONE 2015, 10, e0139179. [Google Scholar] [CrossRef] [PubMed]
- Dekky, B.; Ruff, M.; Bonnier, D.; Legagneux, V.; Théret, N. Proteomic screening identifies the zonula occludens protein ZO-1 as a new partner for ADAM12 in invadopodia-like structures. Oncotarget 2018, 9, 21366–21382. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Liang, L.; Chen, J.; Qiu, N.; Ge, S.; Ji, S.; Shi, T.; Zhen, B.; Liu, M.; Ding, C.; et al. Multidimensional Proteomics Reveals a Role of UHRF2 in the Regulation of Epithelial-Mesenchymal Transition (EMT). Mol. Cell. Proteom. 2016, 15, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Nitschke, A.M.; Xiong, W.; Zhang, Q.; Tang, Y.; Bloch, M.; Elliott, S.; Zhu, Y.; Bazzone, L.; Yu, D.; et al. Proteomic analysis of tumor necrosis factor-α resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008, 10, R105. [Google Scholar] [CrossRef]
- Chen, Y.S.; Mathias, R.A.; Mathivanan, S.; Kapp, E.A.; Moritz, R.L.; Zhu, H.J.; Simpson, R.J. Proteomics profiling of Madin-Darby canine kidney plasma membranes reveals Wnt-5a involvement during oncogenic H-Ras/TGF-β-mediated epithelial-mesenchymal transition. Mol. Cell. Proteom. 2011, 10, M110.001131. [Google Scholar] [CrossRef]
- Grassi, M.L.; Palma, C.S.; Thome, C.H.; Lanfredi, G.P.; Poersch, A.; Faca, V.M. Proteomic analysis of ovarian cancer cells during epithelial-mesenchymal transition (EMT) induced by epidermal growth factor (EGF) reveals mechanisms of cell cycle control. J. Proteom. 2017, 151, 2–11. [Google Scholar] [CrossRef]
- Palma Cde, S.; Grassi, M.L.; Thome, C.H.; Ferreira, G.A.; Albuquerque, D.; Pinto, M.T.; Ferreira Melo, F.U.; Kashima, S.; Covas, D.T.; Pitteri, S.J.; et al. Proteomic Analysis of Epithelial to Mesenchymal Transition (EMT) Reveals Cross-talk between SNAIL and HDAC1 Proteins in Breast Cancer Cells. Mol. Cell. Proteom. 2016, 15, 906–917. [Google Scholar] [CrossRef]
- Silvestrini, V.C.; Thome, C.H.; Albuquerque, D.; de Souza Palma, C.; Ferreira, G.A.; Lanfredi, G.P.; Masson, A.P.; Delsin, L.E.A.; Ferreira, F.U.; de Souza, F.C.; et al. Proteomics analysis reveals the role of ubiquitin specific protease (USP47) in Epithelial to Mesenchymal Transition (EMT) induced by TGFβ2 in breast cells. J. Proteom. 2020, 219, 103734. [Google Scholar] [CrossRef]
- Greening, D.W.; Gopal, S.K.; Mathias, R.A.; Liu, L.; Sheng, J.; Zhu, H.J.; Simpson, R.J. Emerging roles of exosomes during epithelial-mesenchymal transition and cancer progression. Semin. Cell Dev. Biol. 2015, 40, 60–71. [Google Scholar] [CrossRef]
- Xu, R.; Greening, D.W.; Zhu, H.-J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef]
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Couch, Y.; Buzàs, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Théry, C.; et al. A brief history of nearly EV-erything—The rise and rise of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Chen, Y.S.; Wang, B.; Ji, H.; Kapp, E.A.; Moritz, R.L.; Zhu, H.J.; Simpson, R.J. Extracellular remodelling during oncogenic Ras-induced epithelial-mesenchymal transition facilitates MDCK cell migration. J. Proteome Res. 2010, 9, 1007–1019. [Google Scholar] [CrossRef]
- Mathias, R.A.; Wang, B.; Ji, H.; Kapp, E.A.; Moritz, R.L.; Zhu, H.J.; Simpson, R.J. Secretome-based proteomic profiling of Ras-transformed MDCK cells reveals extracellular modulators of epithelial-mesenchymal transition. J. Proteome Res. 2009, 8, 2827–2837. [Google Scholar] [CrossRef]
- Pegoraro, S.; Ros, G.; Piazza, S.; Sommaggio, R.; Ciani, Y.; Rosato, A.; Sgarra, R.; Del Sal, G.; Manfioletti, G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013, 4, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Resmini, G.; Rizzo, S.; Franchin, C.; Zanin, R.; Penzo, C.; Pegoraro, S.; Ciani, Y.; Piazza, S.; Arrigoni, G.; Sgarra, R.; et al. HMGA1 regulates the Plasminogen activation system in the secretome of breast cancer cells. Sci. Rep. 2017, 7, 11768. [Google Scholar] [CrossRef]
- Erin, N.; Ogan, N.; Yerlikaya, A. Secretomes reveal several novel proteins as well as TGF-β1 as the top upstream regulator of metastatic process in breast cancer. Breast Cancer Res. Treat. 2018, 170, 235–250. [Google Scholar] [CrossRef]
- Tauro, B.J.; Mathias, R.A.; Greening, D.W.; Gopal, S.K.; Ji, H.; Kapp, E.A.; Coleman, B.M.; Hill, A.F.; Kusebauch, U.; Hallows, J.L.; et al. Oncogenic H-ras reprograms Madin-Darby canine kidney (MDCK) cell-derived exosomal proteins following epithelial-mesenchymal transition. Mol. Cell. Proteom. 2013, 12, 2148–2159. [Google Scholar] [CrossRef]
- Gopal, S.K.; Greening, D.W.; Hanssen, E.G.; Zhu, H.J.; Simpson, R.J.; Mathias, R.A. Oncogenic epithelial cell-derived exosomes containing Rac1 and PAK2 induce angiogenesis in recipient endothelial cells. Oncotarget 2016, 7, 19709–19722. [Google Scholar] [CrossRef]
- Rai, A.; Fang, H.; Claridge, B.; Simpson, R.J.; Greening, D.W. Proteomic dissection of large extracellular vesicle surfaceome unravels interactive surface platform. J. Extracell. Vesicles 2021, 10, e12164. [Google Scholar] [CrossRef]
- Cvjetkovic, A.; Jang, S.C.; Konečná, B.; Höög, J.L.; Sihlbom, C.; Lässer, C.; Lötvall, J. Detailed Analysis of Protein Topology of Extracellular Vesicles–Evidence of Unconventional Membrane Protein Orientation. Sci. Rep. 2016, 6, 36338. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Nawrocki, A.; Jensen, S.G.; Thorsen, K.; Whitehead, B.; Howard, K.A.; Dyrskjøt, L.; Ørntoft, T.F.; Larsen, M.R.; Ostenfeld, M.S. Quantitative proteomics of fractionated membrane and lumen exosome proteins from isogenic metastatic and nonmetastatic bladder cancer cells reveal differential expression of EMT factors. Proteomics 2014, 14, 699–712. [Google Scholar] [CrossRef]
- Wu, D.; Yan, J.; Shen, X.; Sun, Y.; Thulin, M.; Cai, Y.; Wik, L.; Shen, Q.; Oelrich, J.; Qian, X.; et al. Profiling surface proteins on individual exosomes using a proximity barcoding assay. Nat. Commun. 2019, 10, 3854. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Greening, D.W.; Chen, M.; Rai, A.; Ji, H.; Takahashi, N.; Simpson, R.J. Surfaceome of Exosomes Secreted from the Colorectal Cancer Cell Line SW480: Peripheral and Integral Membrane Proteins Analyzed by Proteolysis and TX114. Proteomics 2019, 19, 1700453. [Google Scholar] [CrossRef] [PubMed]
- Zaborowski, M.P.; Lee, K.; Na, Y.J.; Sammarco, A.; Zhang, X.; Iwanicki, M.; Cheah, P.S.; Lin, H.-Y.; Zinter, M.; Chou, C.-Y. Methods for systematic identification of membrane proteins for specific capture of cancer-derived extracellular vesicles. Cell Rep. 2019, 27, 255–268.e256. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.M.; Gromov, P.; Celis, J.E. Expression of the tumor suppressor protein 14-3-3 sigma is down-regulated in invasive transitional cell carcinomas of the urinary bladder undergoing epithelial-to-mesenchymal transition. Mol. Cell. Proteom. 2004, 3, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Poon, R.T.P.; Lee, N.P.; Yeung, C.; Chan, K.L.; Ng, I.O.L.; Day, P.J.R.; Luk, J.M. Proteomics of Hepatocellular Carcinoma: Serum Vimentin As a Surrogate Marker for Small Tumors (≤2 cm). J. Proteome Res. 2010, 9, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Celis, J.E.; Celis, P.; Palsdottir, H.; Ostergaard, M.; Gromov, P.; Primdahl, H.; Orntoft, T.F.; Wolf, H.; Celis, A.; Gromova, I. Proteomic strategies to reveal tumor heterogeneity among urothelial papillomas. Mol. Cell. Proteom. 2002, 1, 269–279. [Google Scholar] [CrossRef]
- Celis, J.E.; Ostergaard, M.; Basse, B.; Celis, A.; Lauridsen, J.B.; Ratz, G.P.; Andersen, I.; Hein, B.; Wolf, H.; Orntoft, T.F.; et al. Loss of adipocyte-type fatty acid binding protein and other protein biomarkers is associated with progression of human bladder transitional cell carcinomas. Cancer Res. 1996, 56, 4782–4790. [Google Scholar]
- Jacquemier, J.; Ginestier, C.; Rougemont, J.; Bardou, V.J.; Charafe-Jauffret, E.; Geneix, J.; Adélaïde, J.; Koki, A.; Houvenaeghel, G.; Hassoun, J.; et al. Protein expression profiling identifies subclasses of breast cancer and predicts prognosis. Cancer Res. 2005, 65, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Cawthorn, T.R.; Moreno, J.C.; Dharsee, M.; Tran-Thanh, D.; Ackloo, S.; Zhu, P.H.; Sardana, G.; Chen, J.; Kupchak, P.; Jacks, L.M.; et al. Proteomic analyses reveal high expression of decorin and endoplasmin (HSP90B1) are associated with breast cancer metastasis and decreased survival. PLoS ONE 2012, 7, e30992. [Google Scholar] [CrossRef] [PubMed]
- Stroggilos, R.; Mokou, M.; Latosinska, A.; Makridakis, M.; Lygirou, V.; Mavrogeorgis, E.; Drekolias, D.; Frantzi, M.; Mullen, W.; Fragkoulis, C.; et al. Proteome-based classification of Nonmuscle Invasive Bladder Cancer. Int. J. Cancer 2020, 146, 281–294. [Google Scholar] [CrossRef]
- Jézéquel, P.; Campion, L.; Spyratos, F.; Loussouarn, D.; Campone, M.; Guérin-Charbonnel, C.; Joalland, M.-P.; André, J.; Descotes, F.; Grenot, C.; et al. Validation of tumor-associated macrophage ferritin light chain as a prognostic biomarker in node-negative breast cancer tumors: A multicentric 2004 national PHRC study. Int. J. Cancer 2012, 131, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Ricolleau, G.; Charbonnel, C.; Lode, L.; Loussouarn, D.; Joalland, M.P.; Bogumil, R.; Jourdain, S.; Minvielle, S.; Campone, M.; Deporte-Fety, R.; et al. Surface-enhanced laser desorption/ionization time of flight mass spectrometry protein profiling identifies ubiquitin and ferritin light chain as prognostic biomarkers in node-negative breast cancer tumors. Proteomics 2006, 6, 1963–1975. [Google Scholar] [CrossRef]
- Celis, J.E.; Rasmussen, H.H.; Vorum, H.; Madsen, P.; Honoré, B.; Wolf, H.; Orntoft, T.F. Bladder squamous cell carcinomas express psoriasin and externalize it to the urine. J. Urol. 1996, 155, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, M.; Rasmussen, H.H.; Nielsen, H.V.; Vorum, H.; Orntoft, T.F.; Wolf, H.; Celis, J.E. Proteome profiling of bladder squamous cell carcinomas: Identification of markers that define their degree of differentiation. Cancer Res. 1997, 57, 4111–4117. [Google Scholar] [PubMed]
- Hu, X.; Zhang, Y.; Zhang, A.; Li, Y.; Zhu, Z.; Shao, Z.; Zeng, R.; Xu, L.X. Comparative serum proteome analysis of human lymph node negative/positive invasive ductal carcinoma of the breast and benign breast disease controls via label-free semiquantitative shotgun technology. Omics 2009, 13, 291–300. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Johansson, H.J.; Socciarelli, F.; Vacanti, N.M.; Haugen, M.H.; Zhu, Y.; Siavelis, I.; Fernandez-Woodbridge, A.; Aure, M.R.; Sennblad, B.; Vesterlund, M.; et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 2019, 10, 1600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Mun, D.G.; Bhin, J.; Kim, S.; Kim, H.; Jung, J.H.; Jung, Y.; Jang, Y.E.; Park, J.M.; Kim, H.; Jung, Y.; et al. Proteogenomic Characterization of Human Early-Onset Gastric Cancer. Cancer Cell 2019, 35, 111–124.e110. [Google Scholar] [CrossRef] [PubMed]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.W.; Reva, B.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182, 200–225.e235. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, G.; Chakraborty, S.; Das, T.; Boerries, M. Alteration of Proteotranscriptomic Landscape Reveals the Transcriptional Regulatory Circuits Controlling Key-Signaling Pathways and Metabolic Reprogramming During Tumor Evolution. Front. Cell Dev. Biol. 2020, 8, 586479. [Google Scholar] [CrossRef]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Edfors, F.; Danielsson, F.; Hallström, B.M.; Käll, L.; Lundberg, E.; Pontén, F.; Forsström, B.; Uhlén, M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol. 2016, 12, 883. [Google Scholar] [CrossRef]
- Paweletz, C.P.; Charboneau, L.; Bichsel, V.E.; Simone, N.L.; Chen, T.; Gillespie, J.W.; Emmert-Buck, M.R.; Roth, M.J.; Petricoin, I.E.; Liotta, L.A. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 2001, 20, 1981–1989. [Google Scholar] [CrossRef]
- Gulmann, C.; Sheehan, K.M.; Conroy, R.M.; Wulfkuhle, J.D.; Espina, V.; Mullarkey, M.J.; Kay, E.W.; Liotta, L.A.; Petricoin, E.F., 3rd. Quantitative cell signalling analysis reveals down-regulation of MAPK pathway activation in colorectal cancer. J. Pathol. 2009, 218, 514–519. [Google Scholar] [CrossRef]
- Li, S.; Plouffe, B.D.; Belov, A.M.; Ray, S.; Wang, X.; Murthy, S.K.; Karger, B.L.; Ivanov, A.R. An Integrated Platform for Isolation, Processing, and Mass Spectrometry-based Proteomic Profiling of Rare Cells in Whole Blood. Mol. Cell. Proteom. 2015, 14, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Podolak, J.; Zhao, R.; Shukla, A.K.; Moore, R.J.; Thomas, G.V.; Kelly, R.T. Proteome Profiling of 1 to 5 Spiked Circulating Tumor Cells Isolated from Whole Blood Using Immunodensity Enrichment, Laser Capture Microdissection, Nanodroplet Sample Processing, and Ultrasensitive nanoLC-MS. Anal. Chem. 2018, 90, 11756–11759. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.; Brooks, J.; Batis, N.; Khan, N.; El-Asrag, M.; Nankivell, P.; Mehanna, H.; Taylor, G. Feasibility of mass cytometry proteomic characterisation of circulating tumour cells in head and neck squamous cell carcinoma for deep phenotyping. Br. J. Cancer 2023, 129, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Negishi, R.; Yamakawa, H.; Kobayashi, T.; Horikawa, M.; Shimoyama, T.; Koizumi, F.; Sawada, T.; Oboki, K.; Omuro, Y.; Funasaka, C.; et al. Transcriptomic profiling of single circulating tumor cells provides insight into human metastatic gastric cancer. Commun. Biol. 2022, 5, 20. [Google Scholar] [CrossRef]
- Ring, A.; Campo, D.; Porras, T.B.; Kaur, P.; Forte, V.A.; Tripathy, D.; Lu, J.; Kang, I.; Press, M.F.; Jeong, Y.J.; et al. Circulating Tumor Cell Transcriptomics as Biopsy Surrogates in Metastatic Breast Cancer. Ann. Surg. Oncol. 2022, 29, 2882–2894. [Google Scholar] [CrossRef]
- Poonia, S.; Goel, A.; Chawla, S.; Bhattacharya, N.; Rai, P.; Lee, Y.F.; Yap, Y.S.; West, J.; Bhagat, A.A.; Tayal, J.; et al. Marker-free characterization of full-length transcriptomes of single live circulating tumor cells. Genome Res. 2023, 33, 80–95. [Google Scholar] [CrossRef]
- Thiele, J.A.; Pitule, P.; Hicks, J.; Kuhn, P. Single-Cell Analysis of Circulating Tumor Cells. Methods Mol. Biol. 2019, 1908, 243–264. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, F.; Yao, J.; Mao, C.; Zhu, M.; Qian, M.; Hu, J.; Zhong, H.; Zhou, J.; Shi, X.; et al. Single-cell metabolic fingerprints discover a cluster of circulating tumor cells with distinct metastatic potential. Nat. Commun. 2023, 14, 2485. [Google Scholar] [CrossRef]
- Lu, S.; Chang, C.J.; Guan, Y.; Szafer-Glusman, E.; Punnoose, E.; Do, A.; Suttmann, B.; Gagnon, R.; Rodriguez, A.; Landers, M.; et al. Genomic Analysis of Circulating Tumor Cells at the Single-Cell Level. J. Mol. Diagn. 2020, 22, 770–781. [Google Scholar] [CrossRef]
- Kojima, M.; Harada, T.; Fukazawa, T.; Kurihara, S.; Saeki, I.; Takahashi, S.; Hiyama, E. Single-cell DNA and RNA sequencing of circulating tumor cells. Sci. Rep. 2021, 11, 22864. [Google Scholar] [CrossRef]
- Li, M.; Wu, S.; Zhuang, C.; Shi, C.; Gu, L.; Wang, P.; Guo, F.; Wang, Y.; Liu, Z. Metabolomic analysis of circulating tumor cells derived liver metastasis of colorectal cancer. Heliyon 2023, 9, e12515. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Liu, Q.; Liang, D.; Guo, Y.; Liu, G.; Ren, J.; He, Y.; Shan, B. Circulating Tumor Cell and Metabolites as Novel Biomarkers for Early-Stage Lung Cancer Diagnosis. Front. Oncol. 2021, 11, 630672. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yang, X.; Li, Y.; Zhao, P.; Fu, R.; Ren, T.; Hu, P.; Wu, Y.; Yang, H.; Guo, N. Clinical significance of circulating tumor cells and metabolic signatures in lung cancer after surgical removal. J. Transl. Med. 2020, 18, 243. [Google Scholar] [CrossRef] [PubMed]
- Abouleila, Y.; Onidani, K.; Ali, A.; Shoji, H.; Kawai, T.; Lim, C.T.; Kumar, V.; Okaya, S.; Kato, K.; Hiyama, E.; et al. Live single cell mass spectrometry reveals cancer-specific metabolic profiles of circulating tumor cells. Cancer Sci. 2019, 110, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Arbelaiz, A.; Azkargorta, M.; Krawczyk, M.; Santos-Laso, A.; Lapitz, A.; Perugorria, M.J.; Erice, O.; Gonzalez, E.; Jimenez-Aguero, R.; Lacasta, A.; et al. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2017, 66, 1125–1143. [Google Scholar] [CrossRef]
- Shiromizu, T.; Kume, H.; Ishida, M.; Adachi, J.; Kano, M.; Matsubara, H.; Tomonaga, T. Quantitation of putative colorectal cancer biomarker candidates in serum extracellular vesicles by targeted proteomics. Sci. Rep. 2017, 7, 12782. [Google Scholar] [CrossRef]
- Vinik, Y.; Ortega, F.G.; Mills, G.B.; Lu, Y.; Jurkowicz, M.; Halperin, S.; Aharoni, M.; Gutman, M.; Lev, S. Proteomic analysis of circulating extracellular vesicles identifies potential markers of breast cancer progression, recurrence, and response. Sci. Adv. 2020, 6, eaba5714. [Google Scholar] [CrossRef]
- Gurudatt, N.G.; Gwak, H.; Hyun, K.-A.; Jeong, S.-E.; Lee, K.; Park, S.; Chung, M.J.; Kim, S.-E.; Jo, J.H.; Jung, H.-I. Electrochemical detection and analysis of tumor-derived extracellular vesicles to evaluate malignancy of pancreatic cystic neoplasm using integrated microfluidic device. Biosens. Bioelectron. 2023, 226, 115124. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, J.; Zhang, Z.; Wuethrich, A.; Lobb, R.J.; Trau, M. Tracking the EMT-like phenotype switching during targeted therapy in melanoma by analyzing extracellular vesicle phenotypes. Biosens. Bioelectron. 2024, 244, 115819. [Google Scholar] [CrossRef]
- Lee, S.E.; Park, H.Y.; Hur, J.Y.; Kim, H.J.; Kim, I.A.; Kim, W.S.; Lee, K.Y. Genomic profiling of extracellular vesicle-derived DNA from bronchoalveolar lavage fluid of patients with lung adenocarcinoma. Transl. Lung Cancer Res. 2021, 10, 104–116. [Google Scholar] [CrossRef]
- Vitale, S.R.; Helmijr, J.A.; Gerritsen, M.; Coban, H.; van Dessel, L.F.; Beije, N.; van der Vlugt-Daane, M.; Vigneri, P.; Sieuwerts, A.M.; Dits, N.; et al. Detection of tumor-derived extracellular vesicles in plasma from patients with solid cancer. BMC Cancer 2021, 21, 315. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Kasumova, G.G.; Michaud, W.A.; Cintolo-Gonzalez, J.; Díaz-Martínez, M.; Ohmura, J.; Mehta, A.; Chien, I.; Frederick, D.T.; Cohen, S.; et al. Plasma-derived extracellular vesicle analysis and deconvolution enable prediction and tracking of melanoma checkpoint blockade outcome. Sci. Adv. 2020, 6, eabb3461. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Tan, Z.; Zhang, J.; An, M.; Khaykin, V.M.; Cuneo, K.C.; Parikh, N.D.; Lubman, D.M. Sequential Method for Analysis of CTCs and Exosomes from the Same Sample of Patient Blood. ACS Omega 2022, 7, 37581–37588. [Google Scholar] [CrossRef]
- Paul, I.; Bolzan, D.; Youssef, A.; Gagnon, K.A.; Hook, H.; Karemore, G.; Oliphant, M.U.J.; Lin, W.; Liu, Q.; Phanse, S.; et al. Parallelized multidimensional analytic framework applied to mammary epithelial cells uncovers regulatory principles in EMT. Nat. Commun. 2023, 14, 688. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.L.; Balasas, T.; Callaghan, J.; Coombes, R.C.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019, 16, 185–204. [Google Scholar] [CrossRef]
- Fonseca Teixeira, A.; Iaria, J.; Zhu, H.J. Fast Quantitation of TGF-β Signaling Using Adenoviral Reporter. Methods Mol. Biol. 2022, 2488, 13–22. [Google Scholar] [CrossRef]
- Luwor, R.B.; Wang, B.; Nheu, T.V.; Iaria, J.; Tsantikos, E.; Hibbs, M.L.; Sieber, O.M.; Zhu, H.J. New reagents for improved in vitro and in vivo examination of TGF-β signalling. Growth Factors 2011, 29, 211–218. [Google Scholar] [CrossRef]
- Chen, H.; Ware, T.M.B.; Iaria, J.; Zhu, H.J. Live Cell Imaging of the TGF- β/Smad3 Signaling Pathway In Vitro and In Vivo Using an Adenovirus Reporter System. J. Vis. Exp. 2018, 137, e57926. [Google Scholar] [CrossRef]
- Liu, S.; Iaria, J.; Simpson, R.J.; Zhu, H.J. Ras enhances TGF-β signaling by decreasing cellular protein levels of its type II receptor negative regulator SPSB1. Cell Commun. Signal. 2018, 16, 10. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.-H. Signaling networks guiding epithelial–mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.-H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Muthusamy, B.-P.; Derynck, R. The insulin response integrates increased TGF-β signaling through Akt-induced enhancement of cell surface delivery of TGF-β receptors. Sci. Signal. 2015, 8, ra96. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef]
- Hua, W.; Kostidis, S.; Mayboroda, O.; Giera, M.; Hornsveld, M.; ten Dijke, P. Metabolic Reprogramming of Mammary Epithelial Cells during TGF-β-Induced Epithelial-to-Mesenchymal Transition. Metabolites 2021, 11, 626. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca Teixeira, A.; Wu, S.; Luwor, R.; Zhu, H.-J. A New Era of Integration between Multiomics and Spatio-Temporal Analysis for the Translation of EMT towards Clinical Applications in Cancer. Cells 2023, 12, 2740. https://doi.org/10.3390/cells12232740
Fonseca Teixeira A, Wu S, Luwor R, Zhu H-J. A New Era of Integration between Multiomics and Spatio-Temporal Analysis for the Translation of EMT towards Clinical Applications in Cancer. Cells. 2023; 12(23):2740. https://doi.org/10.3390/cells12232740
Chicago/Turabian StyleFonseca Teixeira, Adilson, Siqi Wu, Rodney Luwor, and Hong-Jian Zhu. 2023. "A New Era of Integration between Multiomics and Spatio-Temporal Analysis for the Translation of EMT towards Clinical Applications in Cancer" Cells 12, no. 23: 2740. https://doi.org/10.3390/cells12232740
APA StyleFonseca Teixeira, A., Wu, S., Luwor, R., & Zhu, H. -J. (2023). A New Era of Integration between Multiomics and Spatio-Temporal Analysis for the Translation of EMT towards Clinical Applications in Cancer. Cells, 12(23), 2740. https://doi.org/10.3390/cells12232740