
Novel Molecular Therapies and Genetic Landscape in Selected Rare Diseases with Hematologic Manifestations: A Review of the Literature


Abstract
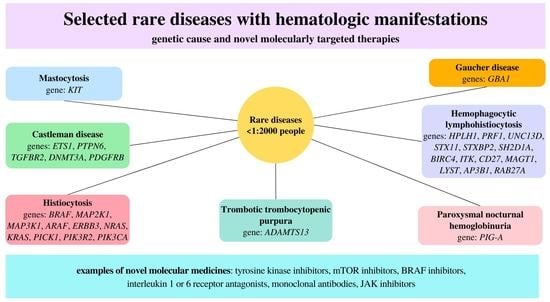
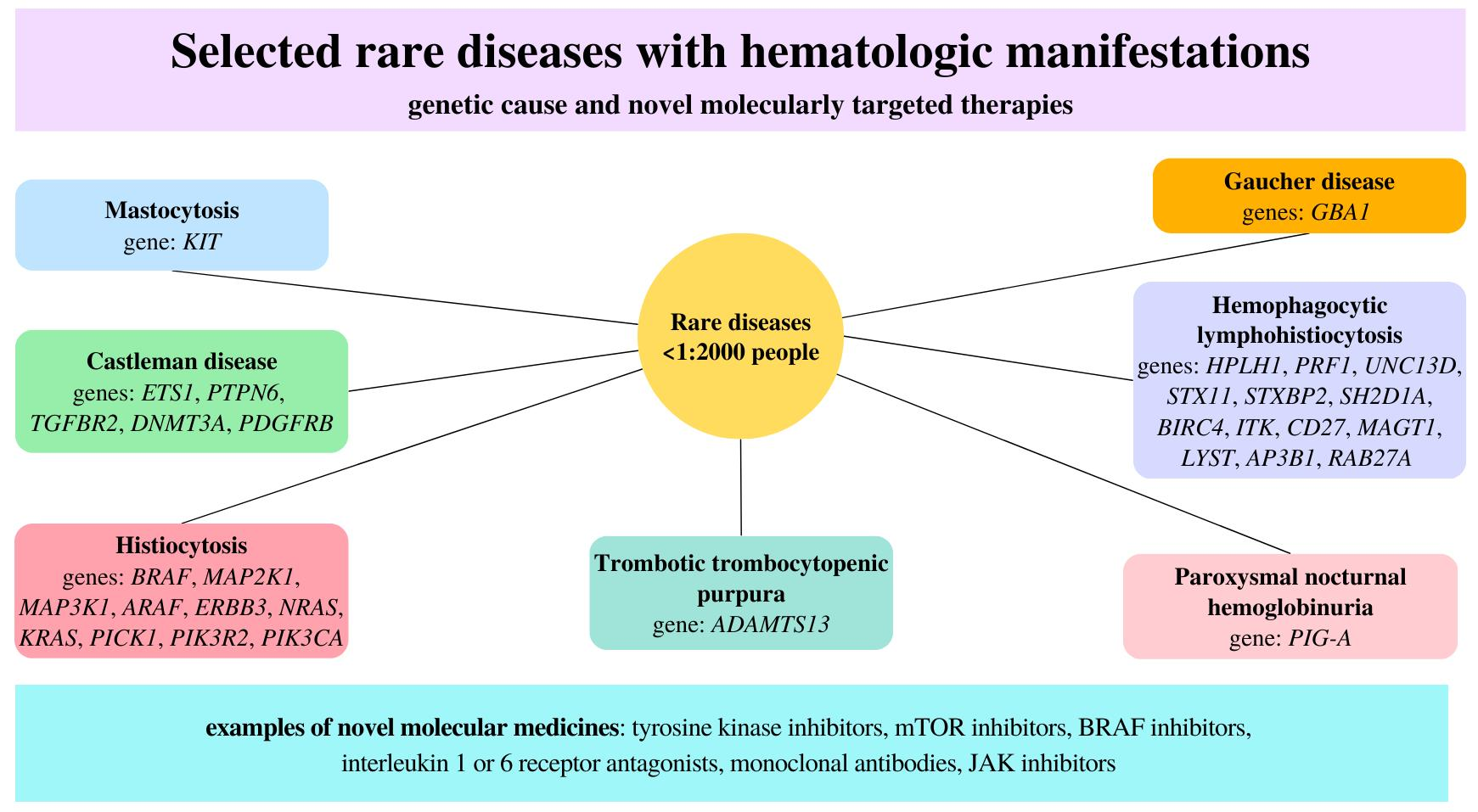
:
1. Introduction
2. Mastocytosis
3. Castleman Disease
4. Langerhans-cell Histiocytosis
5. Thrombotic Thrombocytopenic Purpura
6. Gaucher Disease
7. Hemophagocytic Lymphohistiocytosis
8. Paroxysmal Nocturnal Hemoglobinuria
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AVN | avascular osteonecrosis |
| CD | Castleman disease |
| CDCP1 | CUB domain-containing protein 1 |
| CM | cutaneous mastocytosis |
| CRP | C-reactive protein |
| cTTP | congenital thrombotic thrombocytopenic purpura |
| ERT | enzyme replacement therapy |
| FLH-1 | familial hemophagocytic lymphohistiocytosis type 1 |
| HLH | hemophagocytic lymphohistiocytosis |
| iTTP | immune-mediated thrombotic thrombocytopenic purpura |
| LCH | Langerhans-cell histiocytosis |
| MCD | multicentre Castleman disease |
| NGS | next-generation sequencing |
| PAPP-A | pregnancy-associated plasma protein-A |
| PNH | paroxysmal nocturnal hemoglobinuria |
| SM | systemic mastocytosis |
| TTP | thrombotic thrombocytopenic purpura |
| UCD | unicentric Castleman disease |
References
- Spangenberg, L.; Guecaimburú, R.; Tapié, A.; Vivas, S.; Rodríguez, S.; Graña, M.; Naya, H.; Raggio, V. Novel frameshift mutation in PURA gene causes severe encephalopathy of unclear cause. Mol. Genet. Genom. Med. 2021, 9, e1622. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.V. Pediatric mastocytosis: Recognition and management. Am. J. Clin. Dermatol. 2021, 22, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Van Gysel, D.; DE Maeseneer, H.; Oranje, A.P. Mastocytosis: A comprehensive insight. G Ital. Dermatol. Venereol. 2016, 151, 385–396. [Google Scholar] [PubMed]
- Castells, M.; Metcalfe, D.D.; Escribano, L. Guidelines for the diagnosis and treatment of cutaneous mastocytosis in children. Am. J. Clin. Dermatol. 2011, 12, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.orpha.net/consor/cgi-bin/index.php (accessed on 25 July 2022).
- Gülen, T.; Teufelberger, A.; Ekoff, M.; Westerberg, C.M.; Lyberg, K.; Dahlén, S.E.; Dahlén, B.; Nilsson, G. Distinct plasma biomarkers confirm the diagnosis of mastocytosis and identify increased risk of anaphylaxis. J. Allergy Clin. Immunol. 2021, 148, 889–894. [Google Scholar] [CrossRef]
- Martelli, M.; Monaldi, C.; De Santis, S.; Bruno, S.; Mancini, M.; Cavo, M.; Soverini, S. Recent advances in the molecular biology of systemic mastocytosis: Implications for diagnosis, prognosis, and therapy. Int. J. Mol. Sci. 2020, 21, 3987. [Google Scholar] [CrossRef]
- Carter, M.C.; Metcalfe, D.D.; Clark, A.S.; Wayne, A.S.; Maric, I. Abnormal bone marrow histopathology in paediatric mastocytosis. Br. J. Haematol. 2015, 168, 865–873. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Tzankov, A.; Duncavage, E.; Craig, F.E.; Kelemen, K.; King, R.L.; Orazi, A.; Quintanilla-Martinez, L.; Reichard, K.K.; Rimsza, L.M.; Wang, S.A.; et al. Mastocytosis. Am. J. Clin. Pathol. 2021, 155, 239–266. [Google Scholar] [CrossRef]
- Fett, N.M.; Teng, J.; Longley, B.J. Familial urticaria pigmentosa: Report of a family and review of the role of KIT mutations. Am. J. Dermatopathol. 2013, 35, 113–116. [Google Scholar] [CrossRef]
- De la Sotta, P.; Romero, W.A.; Kramer, D.; Cárdenas, C.; González, S. Cutaneous mastocytosis in twins: Multiple mastocytomas and urticaria pigmentosa in two pairs of monozygotic twins. Pediatr. Dermatol. 2011, 28, 585–587. [Google Scholar] [CrossRef]
- Van den Poel, B.; Kochuyt, A.M.; Del Biondo, E.; Dewaele, B.; Lierman, E.; Tousseyn, T.; de Hertogh, G.; Vandenberghe, P.; Boeckx, N. Highly sensitive assays are mandatory for the differential diagnosis of patients presenting with symptoms of mast cell activation: Diagnostic work-up of 38 patients. Acta Clin. Belg. 2017, 72, 123–129. [Google Scholar] [CrossRef]
- Shibata, Y.; Hirota, S.; Saito, I.; Asahina, A. Diffuse cutaneous mastocytosis: Identification of KIT mutation and long-term follow-up with serum tryptase level. J. Dermatol. 2021, 48, 672–675. [Google Scholar] [CrossRef]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Gilreath, J.A.; Tchertanov, L.; Deininger, M.W. Novel approaches to treating advanced systemic mastocytosis. Clin. Pharmacol. 2019, 11, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Barete, S.; Lortholary, O.; Damaj, G.; Hirsch, I.; Chandesris, M.O.; Elie, C.; Hamidou, M.; Durieu, I.; Suarez, F.; Grosbois, B.; et al. Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Blood 2015, 126, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Dispenzieri, A.; Fajgenbaum, D.C. Overview of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef]
- Szalat, R.; Munshi, N.C. Diagnosis of Castleman disease. Hematol. Oncol. Clin. North Am. 2018, 32, 53–64. [Google Scholar] [CrossRef]
- Wu, D.; Lim, M.S.; Jaffe, E.S. Pathology of Castleman disease. Hematol. Oncol. Clin. North Am. 2018, 32, 37–52. [Google Scholar] [CrossRef]
- Wang, W.; Medeiros, L.J. Castleman disease. Surg. Pathol. Clin. 2019, 12, 849–863. [Google Scholar] [CrossRef]
- Abramson, J.S. Diagnosis and management of Castleman disease. J. Natl. Compr. Canc. Netw. 2019, 17, 1417–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Zhou, C.J.; Jin, M.; Jin, L.; Zhang, R.; Zhang, Y.H. Clinical characteristics of 5 children with Castleman’s disease and review of literature. Zhonghua Er Ke Za Zhi 2010, 48, 625–628. [Google Scholar] [PubMed]
- Fajgenbaum, D.C.; Shilling, D. Castleman disease pathogenesis. Hematol. Oncol. Clin. North Am. 2018, 32, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fan, L.; Li, J. Progress in the diagnosis and treatment of Castleman disease. Zhonghua Xue Ye Xue Za Zhi 2020, 41, 697–700. [Google Scholar] [PubMed]
- Van Rhee, F.; Oksenhendler, E.; Srkalovic, G.; Voorhees, P.; Lim, M.; Dispenzieri, A.; Ide, M.; Parente, S.; Schey, S.; Streetly, M.; et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv. 2020, 4, 6039–6050. [Google Scholar] [CrossRef]
- Murakami, M.; Johkoh, T.; Hayashi, S.; Ohshima, S.; Mizuki, M.; Nakatsuka, S.I.; Tomobe, M.; Kuroyanagi, K.; Nakasone, A.; Nishimoto, N. Clinicopathologic characteristics of 342 patients with multicentric Castleman disease in Japan. Mod. Rheumatol. 2020, 30, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Nagy, A.; Bhaduri, A.; Shahmarvand, N.; Shahryari, J.; Zehnder, J.L.; Warnke, R.A.; Mughal, T.; Ali, S.; Ohgami, R.S. Next-generation sequencing of idiopathic multicentric and unicentric Castleman disease and follicular dendritic cell sarcomas. Blood Adv. 2018, 2, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lan, X.; Li, C.; Zhang, Y.; Wang, Y.; Xue, W.; Lu, L.; Jin, M.; Zhou, Z.; Wang, X.; et al. Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia 2019, 33, 1035–1038. [Google Scholar] [CrossRef] [Green Version]
- Simpson, D. Epidemiology of Castleman disease. Hematol. Oncol. Clin. North Am. 2018, 32, 1–10. [Google Scholar] [CrossRef]
- Available online: https://www.omim.org/ (accessed on 25 July 2022).
- Allen, C.E.; Merad, M.; McClain, K.L. Langerhans-cell histiocytosis. N. Engl. J. Med. 2018, 379, 856–868. [Google Scholar] [CrossRef]
- Néel, A.; Artifoni, M.; Donadieu, J.; Lorillon, G.; Hamidou, M.; Tazi, A. Histiocytose langerhansienne de l’adulte [Langerhans cell histiocytosis in adults]. Rev. Med. Interne 2015, 36, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Galindo, C.; Allen, C.E. Langerhans cell histiocytosis. Blood 2020, 135, 1319–1331. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tojo, A. Langerhans cell histiocytosis in adults: Advances in pathophysiology and treatment. Cancer Sci. 2018, 109, 3707–3713. [Google Scholar] [CrossRef] [Green Version]
- Krooks, J.; Minkov, M.; Weatherall, A.G. Langerhans cell histiocytosis in children: Diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J. Am. Acad. Dermatol. 2018, 78, 1047–1056. [Google Scholar] [CrossRef]
- Haupt, R.; Minkov, M.; Astigarraga, I.; Schäfer, E.; Nanduri, V.; Jubran, R.; Egeler, R.M.; Janka, G.; Micic, D.; Rodriguez-Galindo, C.; et al. Langerhans cell histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr. Blood Cancer 2013, 60, 175–184. [Google Scholar] [CrossRef]
- Bhatia, P.; Singh, M.; Sharma, M.; Sharma, A.; Kakkar, N.; Radhika, S.; Trehan, A.; Bansal, D. BRAF V600E mutation in childhood Langerhans cell histiocytosis correlates with multisystem disease and poor survival. Blood Cells Mol. Dis. 2020, 82, 102356. [Google Scholar] [CrossRef]
- Schönfeld, N.; Dirks, K.; Costabel, U.; Loddenkemper, R.; Wissenschaftliche Arbeitsgemeinschaft für die Therapie von Lungenkrankheiten. A prospective clinical multicentre study on adult pulmonary Langerhans’ cell histiocytosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2012, 29, 132–138. [Google Scholar]
- Uppal, P.; Bothra, M.; Seth, R.; Iyer, V.; Kabra, S.K. Clinical profile of Langerhans Cell Histiocytosis at a tertiary centre: A prospective study. Indian J. Pediatr. 2012, 79, 1463–1467. [Google Scholar] [CrossRef]
- Monsereenusorn, C.; Rodriguez-Galindo, C. Clinical characteristics and treatment of Langerhans cell histiocytosis. Hematol. Oncol. Clin. North Am. 2015, 29, 853–873. [Google Scholar] [CrossRef]
- Su, M.; Gao, Y.J.; Pan, C.; Chen, J.; Tang, J.Y. Outcome of children with Langerhans cell histiocytosis and single-system involvement: A retrospective study at a single center in Shanghai, China. Pediatr. Hematol. Oncol. 2018, 35, 385–392. [Google Scholar] [CrossRef]
- Sukumar, S.; Lämmle, B.; Cataland, S.R. Thrombotic thrombocytopenic purpura: Pathophysiology, diagnosis, and management. J. Clin. Med. 2021, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Yamaguchi, M.; Nishijima, K.; Moriyama, M.; Takakuwa, K.; Enomoto, T. A successfully treated case of an acute presentation of congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome) with decreased ADAMTS13 during late stage of pregnancy. J. Obstet. Gynaecol. Res. 2021, 47, 1892–1897. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.; Podger, L.; Bottomley, C.; Rzepa, E.; Bailey, K.M.A.; Chandler, F. Survival after acute episodes of immune-mediated thrombotic thrombocytopenic purpura (iTTP)—Cognitive functioning and health-related quality of life impact: A descriptive cross-sectional survey of adults living with iTTP in the United Kingdom. Hematology 2021, 26, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, L. Clinical features and gene mutation analysis of congenital thrombotic thrombocytopenic purpura in neonates. Front. Pediatr. 2020, 8, 546248. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Cao, W.; Halkidis, K.; Abdelgawwad, M.S.; Kocher, N.K.; Guillory, B.; Williams, L.A.; Gangaraju, R.; Marques, M.B.; Zheng, X.L. Longitudinal assessments of plasma ADAMTS13 biomarkers predict recurrence of immune thrombotic thrombocytopenic purpura. Blood Adv. 2019, 3, 4177–4186. [Google Scholar] [CrossRef]
- Krogh, A.S.; Waage, A.; Quist-Paulsen, P. Congenital thrombotic thrombocytopenic purpura. Tidsskr Nor Laegeforen 2016, 136, 1452–1457. [Google Scholar] [CrossRef] [Green Version]
- Toret, E.; Demir-Kolsuz, O.; Ozdemir, Z.C.; Bor, O. A case report of congenital thrombotic thrombocytopenic purpura: The peripheral blood smear lights the diagnosis. J. Pediatr. Hematol. Oncol. 2020, 44, e243–e245. [Google Scholar] [CrossRef]
- Kubo, M.; Sakai, K.; Yoshii, Y.; Hayakawa, M.; Matsumoto, M. Rituximab prolongs the time to relapse in patients with immune thrombotic thrombocytopenic purpura: Analysis of off-label use in Japan. Int. J. Hematol. 2020, 112, 764–772. [Google Scholar] [CrossRef]
- Hrdinová, J.; D’Angelo, S.; Graça, N.A.G.; Ercig, B.; Vanhoorelbeke, K.; Veyradier, A.; Voorberg, J.; Coppo, P. Dissecting the pathophysiology of immune thrombotic thrombocytopenic purpura: Interplay between genes and environmental triggers. Haematologica 2018, 103, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Cuker, A.; Cataland, S.R.; Coppo, P.; de la Rubia, J.; Friedman, K.D.; George, J.N.; Knoebl, P.N.; Kremer Hovinga, J.A.; Lämmle, B.; Matsumoto, M.; et al. Redefining outcomes in immune TTP: An international working group consensus report. Blood 2021, 137, 1855–1861. [Google Scholar] [CrossRef]
- Froissart, A.; Buffet, M.; Veyradier, A.; Poullin, P.; Provôt, F.; Malot, S.; Schwarzinger, M.; Galicier, L.; Vanhille, P.; Vernant, J.P.; et al. Efficacy and safety of first-line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a subop-timal response to plasma exchange. Experience of the French Thrombotic Microangiopathies Reference Center. Crit. Care Med. 2012, 40, 104–111. [Google Scholar] [CrossRef]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knöbl, P.; Wu, H.; Artoni, A.; Westwood, J.P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Y.; Stirnemann, J.; Belmatoug, N. La maladie de Gaucher: Quand y penser? [Gaucher disease: A review]. Rev. Med. Interne 2019, 40, 313–322. [Google Scholar] [CrossRef]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. 1), 72–81. [Google Scholar]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef]
- Hughes, D.; Mikosch, P.; Belmatoug, N.; Carubbi, F.; Cox, T.; Goker-Alpan, O.; Kindmark, A.; Mistry, P.; Poll, L.; Weinreb, N.; et al. Gaucher disease in bone: From pathophysiology to practice. J. Bone Miner. Res. 2019, 34, 996–1013. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2017, 120, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Pastores, G. Pharmacological treatment of pediatric Gaucher disease. Exp. Rev. Clin. Pharmacol. 2018, 11, 1183–1194. [Google Scholar] [CrossRef]
- Lam, M.T.; Coppola, S.; Krumbach, O.H.F.; Prencipe, G.; Insalaco, A.; Cifaldi, C.; Brigida, I.; Zara, E.; Scala, S.; Di Cesare, S.; et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J. Exp. Med. 2019, 216, 2778–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burak, N.; Jan, N.; Kessler, J.; Oei, E.; Patel, P.; Feldman, S. Diagnosis of GATA2 deficiency in a young woman with hemophagocytic lymphohistiocytosis triggered by acute systemic cytomegalovirus infection. Am. J. Case Rep. 2021, 22, e927087. [Google Scholar] [CrossRef] [PubMed]
- Cetica, V.; Sieni, E.; Pende, D.; Danesino, C.; De Fusco, C.; Locatelli, F.; Micalizzi, C.; Putti, M.C.; Biondi, A.; Fagioli, F.; et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J. Allergy Clin. Immunol. 2016, 137, 188–196.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinowska, I.; Machaczka, M.; Popko, K.; Siwicka, A.; Salamonowicz, M.; Nasiłowska-Adamska, B. Hemophagocytic syndrome in children and adults. Arch. Immunol. Ther. Exp. 2014, 62, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, S.J.; Tasian, S.K.; Bassiri, H.; Fisher, B.T.; Atalla, J.; Patel, R.; Romberg, N.; Lambert, M.P.; Paessler, M.; Behrens, E.J.; et al. Diagnostic challenges in pediatric hemophagocytic lymphohistiocytosis. J. Clin. Immunol. 2021, 41, 1213–1218. [Google Scholar] [CrossRef]
- Shabrish, S.; Kelkar, M.; Yadav, R.M.; Bargir, U.A.; Gupta, M.; Dalvi, A.; Aluri, J.; Kulkarni, M.; Shinde, S.; Sawant-Desai, S.; et al. The spectrum of clinical, immunological, and molecular findings in familial hemophagocytic lymphohistiocytosis: Experience from India. Front Immunol. 2021, 12, 612583. [Google Scholar] [CrossRef]
- Abughanimeh, O.; Qasrawi, A.; Abu Ghanimeh, M. Hemophagocytic lymphohistiocytosis complicating systemic sarcoidosis. Cureus 2018, 10, e2838. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, C.A. Comparing hemophagocytic lymphohistiocytosis in pediatric and adult patients. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 405–413. [Google Scholar] [CrossRef]
- Locatelli, F.; Jordan, M.B.; Allen, C.; Cesaro, S.; Rizzari, C.; Rao, A.; Degar, B.; Garrington, T.P.; Sevilla, J.; Putti, M.C.; et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N. Engl. J. Med. 2020, 382, 1811–1822. [Google Scholar] [CrossRef]
- Hansen, S.; Alduaij, W.; Biggs, C.M.; Belga, S.; Luecke, K.; Merkeley, H.; Chen, L.Y.C. Ruxolitinib as adjunctive therapy for secondary hemophagocytic lymphohistiocytosis: A case series. Eur. J. Haematol. 2021, 106, 654–661. [Google Scholar] [CrossRef]
- Farooq, Q.; Saleem, M.W.; Khan, Z.U.; Hadi, N. Paroxysmal nocturnal hemoglobinuria: A diagnostic “zero-sum-game”. Cureus 2020, 12, e11956. [Google Scholar] [CrossRef]
- Cançado, R.D.; Araújo, A.D.S.; Sandes, A.F.; Arrais, C.; Lobo, C.L.C.; Figueiredo, M.S.; Gualandro, S.F.M.; Saad, S.T.O.; Costa, F.F. Consensus statement for diagnosis and treatment of paroxysmal nocturnal haemoglobinuria. Hematol. Transfus. Cell Ther. 2020, 43, 341–348. [Google Scholar] [CrossRef]
- Henderson, C.; Lo, M.; Massey, G. Pediatric paroxysmal nocturnal hemoglobinuria presenting as acute kidney injury. J. Pediatr. Hematol Oncol. 2021, 43, e543–e545. [Google Scholar] [CrossRef]
- Fattizzo, B.; Serpenti, F.; Giannotta, J.A.; Barcellini, W. Difficult cases of paroxysmal nocturnal hemoglobinuria: Diagnosis and therapeutic novelties. J. Clin. Med. 2021, 10, 948. [Google Scholar] [CrossRef]
- Jeong, D.; Park, H.S.; Kim, S.M.; Im, K.; Yun, J.; Lee, Y.E.; Ryu, S.; Ahn, Y.O.; Yoon, S.S.; Lee, D.S. Ultradeep sequencing analysis of paroxysmal nocturnal hemoglobinuria clones detected by flow cytometry: PIG mutation in small PNH clones. Am. J. Clin. Pathol. 2021, 156, 72–85. [Google Scholar] [CrossRef]
- Zhou, S.; Dong, X.; Chen, C.; Ma, L.; Wu, Y.; Zhou, Y.; Cui, Y. Efficacy and safety of eculizumab for paroxysmal nocturnal hemoglobinuria: A systematic review and meta-analysis. J Pediatr. Hematol. Oncol. 2021, 43, 203–210. [Google Scholar] [CrossRef]
- Hillmen, P.; Szer, J.; Weitz, I.; Röth, A.; Höchsmann, B.; Panse, J.; Usuki, K.; Griffin, M.; Kiladjian, J.J.; de Castro, C.; et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2021, 384, 1028–1037. [Google Scholar] [CrossRef]
- Kulasekararaj, A.G.; Hill, A.; Langemeijer, S.; Wells, R.; González Fernández, F.A.; Gaya, A.; Ojeda Gutierrez, E.; Piatek, C.I.; Mitchell, L.; Usuki, K.; et al. One-year outcomes from a phase 3 randomized trial of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria who received prior eculizumab. Eur. J. Haematol. 2021, 106, 389–397. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Diseases | Gene |
|---|---|
| Mastocytosis | KIT |
| Castleman disease | ETS1, PTPN6, TGFBR2, DNMT3A, PDGFRB |
| Histiocytosis | BRAF, MAP2K1, MAP3K1, ARAF, ERBB3, NRAS, KRAS, PICK1, PIK3R2, PIK3CA |
| Trombotic trombocytopenic purpura | ADAMTS13 |
| Gaucher disease | GBA1 |
| Hemophagocytic lymphohistiocytosis | HPLH1, PRF1, UNC13D, STX11, STXBP2, SH2D1A, BIRC4, ITK, CD27, MAGT1, LYST, AP3B1, RAB27A |
| Paroxysmal nocturnal hemoglobinuria | PIG-A |
| Diseases | Novel Treatment |
|---|---|
| Mastocytosis | leukotriene antagonists, H1 and H2 antihistamines, cromolyn sodium, corticosteroids, methoxypsoralen therapy with long-wave psoralen plus ultraviolet A, midostaurin, imatinib, nilotinib, dasatinib, masitinib, avapritinib, ripretinib, cladribine |
| Castleman disease | corticosteroids, rituximab, thalidomide, lenalidomide, bortezomib, cyclosporine, sirolimus, interferon, antiretroviral therapy |
| Langerhans-cell histiocytosis | vemurafenib, dabrafenib, imatinib |
| Trombotic trombocytopenic purpura | therapeutic plasma exchange with fresh frozen plasma replacement, corticosteroids, cyclosporine A, mycophenolate mofetil, rituximab, bortezomib, caplacizumab |
| Gaucher disease | enzyme replacement therapy, imiglucerase, velaglucerase, taliglucerase, substrate reduction therapy, miglustat, eliglustat |
| Hemophagocytic lymphohistiocytosis | corticosteroids, etoposide, cyclosporin, emapalumab, anakinra, ruxolitinib, tofacitnib, baricitinib, itacitinib |
| Paroxysmal nocturnal hemoglobinuria | anti-thrombosis prophylaxis, blood transfusion, allogeneic bone marrow transplantation, eculizumab, pegcetacoplan |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ręka, G.; Stefaniak, M.; Lejman, M. Novel Molecular Therapies and Genetic Landscape in Selected Rare Diseases with Hematologic Manifestations: A Review of the Literature. Cells 2023, 12, 449. https://doi.org/10.3390/cells12030449
Ręka G, Stefaniak M, Lejman M. Novel Molecular Therapies and Genetic Landscape in Selected Rare Diseases with Hematologic Manifestations: A Review of the Literature. Cells. 2023; 12(3):449. https://doi.org/10.3390/cells12030449
Chicago/Turabian StyleRęka, Gabriela, Martyna Stefaniak, and Monika Lejman. 2023. "Novel Molecular Therapies and Genetic Landscape in Selected Rare Diseases with Hematologic Manifestations: A Review of the Literature" Cells 12, no. 3: 449. https://doi.org/10.3390/cells12030449
APA StyleRęka, G., Stefaniak, M., & Lejman, M. (2023). Novel Molecular Therapies and Genetic Landscape in Selected Rare Diseases with Hematologic Manifestations: A Review of the Literature. Cells, 12(3), 449. https://doi.org/10.3390/cells12030449