

Peroxisome Proliferator-Activated Receptor-Targeted Therapies: Challenges upon Infectious Diseases


Abstract
:1. Introduction
2. Overview of PPARs
2.1. Molecular Characteristics of PPARs
2.1.1. Roles of PPARα
2.1.2. Roles of PPARβ/δ
2.1.3. Roles of PPARγ
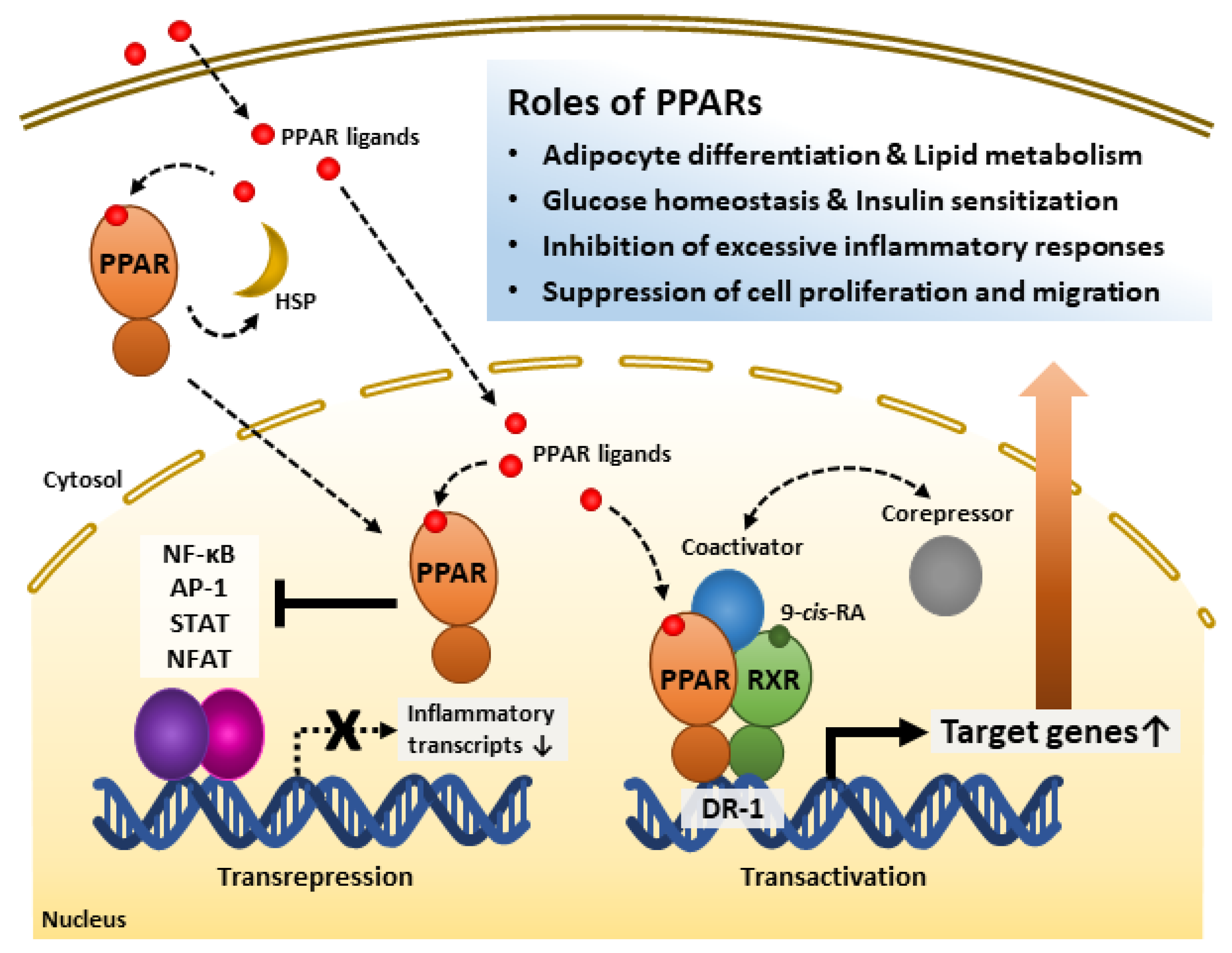
2.2. Regulatory Mechanisms of PPARs
3. PPARs and Viral Infections
3.1. PPARs and Respiratory Viral Infections
3.2. PPARs and Virus-Related Inflammation
3.3. PPARs and Hepatitis Virus Infection
4. PPARs and Bacterial Infections
4.1. PPARs and Post-Influenza Bacterial Infections
4.2. PPARs in Bacterial Infections
4.3. PPARs and Mycobacterial Infections
5. PPARs and Parasitic Infections
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Toobian, D.; Ghosh, P.; Katkar, G.D. Parsing the Role of PPARs in Macrophage Processes. Front. Immunol. 2021, 12, 783780. [Google Scholar] [CrossRef]
- Schupp, M.; Lazar, M.A. Endogenous ligands for nuclear receptors: Digging deeper. J. Biol. Chem. 2010, 285, 40409–40415. [Google Scholar] [CrossRef] [Green Version]
- Lamas Bervejillo, M.; Ferreira, A.M. Understanding Peroxisome Proliferator-Activated Receptors: From the Structure to the Regulatory Actions on Metabolism. Adv. Exp. Med. Biol. 2019, 1127, 39–57. [Google Scholar] [CrossRef]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-Regulated Gene Expression. PPAR Res. 2010, 2010, 250126. [Google Scholar] [CrossRef] [Green Version]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications--a review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhang, T.; Han, H. PPARα: A potential therapeutic target of cholestasis. Front. Pharmacol. 2022, 13, 916866. [Google Scholar] [CrossRef]
- Bordet, R.; Ouk, T.; Petrault, O.; Gelé, P.; Gautier, S.; Laprais, M.; Deplanque, D.; Duriez, P.; Staels, B.; Fruchart, J.C.; et al. PPAR: A new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochem. Soc. Trans. 2006, 34, 1341–1346. [Google Scholar] [CrossRef] [Green Version]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and PPAR gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res. 2015, 2015, 549691. [Google Scholar] [CrossRef] [Green Version]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Derosa, G.; Sahebkar, A.; Maffioli, P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J. Cell. Physiol. 2018, 233, 153–161. [Google Scholar] [CrossRef]
- Diskin, C.; Ryan, T.A.J.; O’Neill, L.A.J. Modification of Proteins by Metabolites in Immunity. Immunity 2021, 54, 19–31. [Google Scholar] [CrossRef]
- Haas, R.; Cucchi, D.; Smith, J.; Pucino, V.; Macdougall, C.E.; Mauro, C. Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem. Sci. 2016, 41, 460–471. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gómez de Las Heras, M.M.; Gabandé-Rodríguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Okumoto, K.; Tamura, S.; Honsho, M.; Fujiki, Y. Peroxisome: Metabolic Functions and Biogenesis. Adv. Exp. Med. Biol. 2020, 1299, 3–17. [Google Scholar] [CrossRef]
- Reddy, J.K.; Krishnakantha, T.P.; Azarnoff, D.L.; Moody, D.E. 1-methyl-4piperidyl-bis (P-chlorophenoxy) acetate: A new hypolipidemic peroxisome proliferator. Res. Commun. Chem. Pathol. Pharmacol. 1975, 10, 589–592. [Google Scholar]
- Reddy, J.K.; Lalwai, N.D. Carcinogenesis by hepatic peroxisome proliferators: Evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. Crit. Rev. Toxicol. 1983, 12, 1–58. [Google Scholar] [CrossRef]
- Reddy, J.K.; Goel, S.K.; Nemali, M.R.; Carrino, J.J.; Laffler, T.G.; Reddy, M.K.; Sperbeck, S.J.; Osumi, T.; Hashimoto, T.; Lalwani, N.D.; et al. Transcription regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc. Natl. Acad. Sci. USA 1986, 83, 1747–1751. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, J.P.; Song, B.J.; Huberman, E.; Gonzalez, F.J. Isolation, complementary DNA sequence, and regulation of rat hepatic lauric acid omega-hydroxylase (cytochrome P-450LA omega). Identification of a new cytochrome P-450 gene family. J. Biol. Chem. 1987, 262, 801–810. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Zhu, Y.; Alvares, K.; Huang, Q.; Rao, M.S.; Reddy, J.K. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J. Biol. Chem. 1993, 268, 26817–26820. [Google Scholar] [CrossRef]
- Göttlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef] [Green Version]
- Rolls, B.J.; Hammer, V.A. Fat, carbohydrate, and the regulation of energy intake. Am. J. Clin. Nutr. 1995, 62, 1086s–1095s. [Google Scholar] [CrossRef]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar]
- Schmidt, A.; Endo, N.; Rutledge, S.J.; Vogel, R.; Shinar, D.; Rodan, G.A. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol. Endocrinol. 1992, 6, 1634–1641. [Google Scholar] [CrossRef] [Green Version]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef]
- Sher, T.; Yi, H.F.; McBride, O.W.; Gonzalez, F.J. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry 1993, 32, 5598–5604. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Qi, J.S.; Desai-Yajnik, V.; Greene, M.E.; Raaka, B.M.; Samuels, H.H. The ligand-binding domains of the thyroid hormone/retinoid receptor gene subfamily function in vivo to mediate heterodimerization, gene silencing, and transactivation. Mol. Cell. Biol. 1995, 15, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef]
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARγ signaling and emerging opportunities for improved therapeutics. Pharmacol. Res. 2016, 111, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Bain, D.L.; Heneghan, A.F.; Connaghan-Jones, K.D.; Miura, M.T. Nuclear receptor structure: Implications for function. Annu. Rev. Physiol. 2007, 69, 201–220. [Google Scholar] [CrossRef]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [Green Version]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [Green Version]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.N.; Hsu, M.H.; Griffin, H.J.; Johnson, E.F. Novel sequence determinants in peroxisome proliferator signaling. J. Biol. Chem. 1995, 270, 16114–16121. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Lazar, M.A. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 1999, 402, 93–96. [Google Scholar] [CrossRef]
- Poulsen, L.; Siersbæk, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef]
- Chan, L.S.; Wells, R.A. Cross-Talk between PPARs and the Partners of RXR: A Molecular Perspective. PPAR Res. 2009, 2009, 925309. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Delerive, P.; Gervois, P.; Fruchart, J.C.; Staels, B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J. Biol. Chem. 2000, 275, 36703–36707. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 1998, 393, 790–793. [Google Scholar] [CrossRef]
- Madej, A.; Okopien, B.; Kowalski, J.; Zielinski, M.; Wysocki, J.; Szygula, B.; Kalina, Z.; Herman, Z.S. Effects of fenofibrate on plasma cytokine concentrations in patients with atherosclerosis and hyperlipoproteinemia IIb. Int. J. Clin. Pharmacol. Ther. 1998, 36, 345–349. [Google Scholar]
- Delerive, P.; Martin-Nizard, F.; Chinetti, G.; Trottein, F.; Fruchart, J.C.; Najib, J.; Duriez, P.; Staels, B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ. Res. 1999, 85, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Fougerat, A.; Schoiswohl, G.; Polizzi, A.; Régnier, M.; Wagner, C.; Smati, S.; Fougeray, T.; Lippi, Y.; Lasserre, F.; Raho, I.; et al. ATGL-dependent white adipose tissue lipolysis controls hepatocyte PPARα activity. Cell Rep. 2022, 39, 110910. [Google Scholar] [CrossRef]
- Wang, A.; Huen, S.C.; Luan, H.H.; Yu, S.; Zhang, C.; Gallezot, J.D.; Booth, C.J.; Medzhitov, R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 2016, 166, 1512–1525.e1512. [Google Scholar] [CrossRef] [Green Version]
- Huen, S.C.; Wang, A.; Feola, K.; Desrouleaux, R.; Luan, H.H.; Hogg, R.; Zhang, C.; Zhang, Q.J.; Liu, Z.P.; Medzhitov, R. Hepatic FGF21 preserves thermoregulation and cardiovascular function during bacterial inflammation. J. Exp. Med. 2021, 218, e20202151. [Google Scholar] [CrossRef]
- Paumelle, R.; Haas, J.T.; Hennuyer, N.; Baugé, E.; Deleye, Y.; Mesotten, D.; Langouche, L.; Vanhoutte, J.; Cudejko, C.; Wouters, K.; et al. Hepatic PPARα is critical in the metabolic adaptation to sepsis. J. Hepatol. 2019, 70, 963–973. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Müller, R. PPARβ/δ in human cancer. Biochimie 2017, 136, 90–99. [Google Scholar] [CrossRef]
- Yao, P.L.; Chen, L.; Dobrzański, T.P.; Zhu, B.; Kang, B.H.; Müller, R.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-β/δ inhibits human neuroblastoma cell tumorigenesis by inducing p53- and SOX2-mediated cell differentiation. Mol. Carcinog. 2017, 56, 1472–1483. [Google Scholar] [CrossRef]
- Foreman, J.E.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) inhibits cell growth in a mouse mammary gland cancer cell line. Cancer Lett. 2010, 288, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Genini, D.; Garcia-Escudero, R.; Carbone, G.M.; Catapano, C.V. Transcriptional and Non-Transcriptional Functions of PPARβ/δ in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e46009. [Google Scholar] [CrossRef]
- Pedchenko, T.V.; Gonzalez, A.L.; Wang, D.; DuBois, R.N.; Massion, P.P. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am. J. Respir. Cell Mol. Biol. 2008, 39, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.D.; Benchetrit, M.; Bianchini, L.; Michiels, J.F.; Wagner, N. Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) is highly expressed in liposarcoma and promotes migration and proliferation. J. Pathol. 2011, 224, 575–588. [Google Scholar] [CrossRef]
- Bishop-Bailey, D. PPARs and angiogenesis. Biochem. Soc. Trans. 2011, 39, 1601–1605. [Google Scholar] [CrossRef]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef]
- Kostadinova, R.; Wahli, W.; Michalik, L. PPARs in diseases: Control mechanisms of inflammation. Curr. Med. Chem. 2005, 12, 2995–3009. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Chinetti, G.; Griglio, S.; Antonucci, M.; Torra, I.P.; Delerive, P.; Majd, Z.; Fruchart, J.C.; Chapman, J.; Najib, J.; Staels, B. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 1998, 273, 25573–25580. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Li, R.; Jiang, J.; Cai, B.; Gao, J.; Le, K.; Zhang, F.; Chen, S.; Liu, P. Activation of peroxisome proliferator-activated receptor gamma inhibits endothelin-1-induced cardiac hypertrophy via the calcineurin/NFAT signaling pathway. Mol. Cell. Biochem. 2008, 317, 189–196. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα Ligand-Binding Domain Structures with Endogenous Fatty Acids and Fibrates. iScience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef]
- Aagaard, M.M.; Siersbæk, R.; Mandrup, S. Molecular basis for gene-specific transactivation by nuclear receptors. Biochim. Biophys. Acta 2011, 1812, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Sumanasekera, W.K.; Tien, E.S.; Davis, J.W., 2nd; Turpey, R.; Perdew, G.H.; Vanden Heuvel, J.P. Heat shock protein-90 (Hsp90) acts as a repressor of peroxisome proliferator-activated receptor-alpha (PPARalpha) and PPARbeta activity. Biochemistry 2003, 42, 10726–10735. [Google Scholar] [CrossRef]
- Wheeler, M.C.; Gekakis, N. Hsp90 modulates PPARγ activity in a mouse model of nonalcoholic fatty liver disease. J. Lipid Res. 2014, 55, 1702–1710. [Google Scholar] [CrossRef] [Green Version]
- Layrolle, P.; Payoux, P.; Chavanas, S. PPAR Gamma and Viral Infections of the Brain. Int. J. Mol. Sci. 2021, 22, 8876. [Google Scholar] [CrossRef]
- Bassaganya-Riera, J.; Song, R.; Roberts, P.C.; Hontecillas, R. PPAR-gamma activation as an anti-inflammatory therapy for respiratory virus infections. Viral Immunol. 2010, 23, 343–352. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, B.; Cheon, I.S.; Goplen, N.P.; Jiang, L.; Zhang, R.; Peebles, R.S.; Mack, M.; Kaplan, M.H.; Limper, A.H.; et al. PPAR-γ in Macrophages Limits Pulmonary Inflammation and Promotes Host Recovery following Respiratory Viral Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhou, Y.H.; Yang, Z.Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Goplen, N.P.; Zhu, B.; Cheon, I.S.; Son, Y.; Wang, Z.; Li, C.; Dai, Q.; Jiang, L.; Xiang, M.; et al. Macrophage PPAR-γ suppresses long-term lung fibrotic sequelae following acute influenza infection. PLoS ONE 2019, 14, e0223430. [Google Scholar] [CrossRef] [Green Version]
- Bei, Y.; Tia, B.; Li, Y.; Guo, Y.; Deng, S.; Huang, R.; Zeng, H.; Li, R.; Wang, G.F.; Dai, J. Anti-influenza A Virus Effects and Mechanisms of Emodin and Its Analogs via Regulating PPARα/γ-AMPK-SIRT1 Pathway and Fatty Acid Metabolism. Biomed. Res. Int. 2021, 2021, 9066938. [Google Scholar] [CrossRef]
- Heffernan, K.S.; Ranadive, S.M.; Jae, S.Y. Exercise as medicine for COVID-19: On PPAR with emerging pharmacotherapy. Med. Hypotheses 2020, 143, 110197. [Google Scholar] [CrossRef]
- Dias, S.S.G.; Soares, V.C.; Ferreira, A.C.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Teixeira, L.; Nunes da Silva, M.A.; Barreto, E.; Mattos, M.; et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020, 16, e1009127. [Google Scholar] [CrossRef]
- Alkhayyat, S.S.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; El-Bouseary, M.M.; AboKamer, A.M.; Batiha, G.E.; Simal-Gandara, J. Fenofibrate for COVID-19 and related complications as an approach to improve treatment outcomes: The missed key for Holy Grail. Inflamm. Res. 2022, 71, 1159–1167. [Google Scholar] [CrossRef]
- Jagat, J.M.; Kalyan, K.G.; Subir, R. Use of pioglitazone in people with type 2 diabetes mellitus with coronavirus disease 2019 (COVID-19): Boon or bane? Diabetol. Metab. Syndr. 2020, 14, 829–831. [Google Scholar] [CrossRef]
- Nagarkatti, P.; Miranda, K.; Nagarkatti, M. Use of Cannabinoids to Treat Acute Respiratory Distress Syndrome and Cytokine Storm Associated with Coronavirus Disease-2019. Front. Pharmacol. 2020, 11, 589438. [Google Scholar] [CrossRef]
- Corpetti, C.; Del Re, A.; Seguella, L.; Palenca, I.; Rurgo, S.; De Conno, B.; Pesce, M.; Sarnelli, G.; Esposito, G. Cannabidiol inhibits SARS-Cov-2 spike (S) protein-induced cytotoxicity and inflammation through a PPARγ-dependent TLR4/NLRP3/Caspase-1 signaling suppression in Caco-2 cell line. Phytother. Res. 2021, 35, 6893–6903. [Google Scholar] [CrossRef]
- Francisqueti-Ferron, F.V.; Garcia, J.L.; Ferron, A.J.T.; Nakandakare-Maia, E.T.; Gregolin, C.S.; Silva, J.; Dos Santos, K.C.; Lo, Â.T.C.; Siqueira, J.S.; de Mattei, L.; et al. Gamma-oryzanol as a potential modulator of oxidative stress and inflammation via PPAR-y in adipose tissue: A hypothetical therapeutic for cytokine storm in COVID-19? Mol. Cell. Endocrinol. 2021, 520, 111095. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Lyroni, K.; Aznaourova, M.; Tseliou, M.; Al-Anazi, M.R.; Al-Ahdal, M.N.; Alkahtani, S.; Sourvinos, G.; Tsatsanis, C. Middle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPARγ. Oncotarget 2017, 8, 9053–9066. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Mamidi, P.; Ghosh, S.; Keshry, S.S.; Mahish, C.; Pani, S.S.; Laha, E.; Ray, A.; Datey, A.; Chatterjee, S.; et al. Telmisartan Restricts Chikungunya Virus Infection In Vitro and In Vivo through the AT1/PPAR-γ/MAPKs Pathways. Antimicrob. Agents Chemother. 2022, 66, e0148921. [Google Scholar] [CrossRef]
- Omeragic, A.; Hoque, M.T.; Choi, U.Y.; Bendayan, R. Peroxisome proliferator-activated receptor-gamma: Potential molecular therapeutic target for HIV-1-associated brain inflammation. J. Neuroinflamm. 2017, 14, 183. [Google Scholar] [CrossRef]
- Jiang, H.; Cheng, S.T.; Ren, J.H.; Ren, F.; Yu, H.B.; Wang, Q.; Huang, A.L.; Chen, J. SIRT6 Inhibitor, OSS_128167 Restricts Hepatitis B Virus Transcription and Replication Through Targeting Transcription Factor Peroxisome Proliferator-Activated Receptors α. Front. Pharmacol. 2019, 10, 1270. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.M.; Sun, H.Y.; Chiu, W.T.; Su, H.C.; Chien, Y.C.; Chong, L.W.; Chang, H.C.; Bai, C.H.; Young, K.C.; Tsao, C.W. Calcitriol Inhibits HCV Infection via Blockade of Activation of PPAR and Interference with Endoplasmic Reticulum-Associated Degradation. Viruses 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Mansfield, J.; Fan, R.; MacLean, A.; Li, J.; Mohan, M. miR-130a and miR-212 Disrupt the Intestinal Epithelial Barrier through Modulation of PPARγ and Occludin Expression in Chronic Simian Immunodeficiency Virus-Infected Rhesus Macaques. J. Immunol. 2018, 200, 2677–2689. [Google Scholar] [CrossRef] [Green Version]
- Crakes, K.R.; Santos Rocha, C.; Grishina, I.; Hirao, L.A.; Napoli, E.; Gaulke, C.A.; Fenton, A.; Datta, S.; Arredondo, J.; Marco, M.L.; et al. PPARα-targeted mitochondrial bioenergetics mediate repair of intestinal barriers at the host-microbe intersection during SIV infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24819–24829. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Huang, C.H.; Wang, W.H.; Tenhunen, J.; Hung, L.C.; Lin, C.C.; Chen, Y.C.; Chen, Y.H.; Liao, W.T. Mono-(2-ethylhexyl) phthalate Promotes Dengue Virus Infection by Decreasing IL-23-Mediated Antiviral Responses. Front. Immunol. 2021, 12, 599345. [Google Scholar] [CrossRef]
- Mulkey, S.B.; Arroyave-Wessel, M.; Peyton, C.; Bulas, D.I.; Fourzali, Y.; Jiang, J.; Russo, S.; McCarter, R.; Msall, M.E.; du Plessis, A.J.; et al. Neurodevelopmental Abnormalities in Children with In Utero Zika Virus Exposure Without Congenital Zika Syndrome. JAMA Pediatr. 2020, 174, 269–276. [Google Scholar] [CrossRef]
- Thulasi Raman, S.N.; Latreille, E.; Gao, J.; Zhang, W.; Wu, J.; Russell, M.S.; Walrond, L.; Cyr, T.; Lavoie, J.R.; Safronetz, D.; et al. Dysregulation of Ephrin receptor and PPAR signaling pathways in neural progenitor cells infected by Zika virus. Emerg. Microbes Infect. 2020, 9, 2046–2060. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, Y.; Feng, S.; Ishida, Y.; Chiu, T.P.; Saito, T.; Wang, S.; Ann, D.K.; Ou, J.J. Macrophages activated by hepatitis B virus have distinct metabolic profiles and suppress the virus via IL-1β to downregulate PPARα and FOXO3. Cell Rep. 2022, 38, 110284. [Google Scholar] [CrossRef]
- Du, L.; Ma, Y.; Liu, M.; Yan, L.; Tang, H. Peroxisome Proliferators Activated Receptor (PPAR) agonists activate hepatitis B virus replication in vivo. Virol. J. 2017, 14, 96. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Suda, G.; Yamamoto, Y.; Furuya, K.; Baba, M.; Nakamura, A.; Miyoshi, H.; Kimura, M.; Maehara, O.; Yamada, R.; et al. Tenofovir-disoproxil-fumarate modulates lipid metabolism via hepatic CD36/PPAR-alpha activation in hepatitis B virus infection. J. Gastroenterol. 2021, 56, 168–180. [Google Scholar] [CrossRef]
- Goldwasser, J.; Cohen, P.Y.; Lin, W.; Kitsberg, D.; Balaguer, P.; Polyak, S.J.; Chung, R.T.; Yarmush, M.L.; Nahmias, Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J. Hepatol. 2011, 55, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.C.; Suen, R.; Treuting, P.M.; Armando, A.; Lucarelli, R.; Gorrochotegui-Escalante, N.; Diercks, A.H.; Quehenberger, O.; Dennis, E.A.; Aderem, A.; et al. PPARα exacerbates necroptosis, leading to increased mortality in postinfluenza bacterial superinfection. Proc. Natl. Acad. Sci. USA 2020, 117, 15789–15798. [Google Scholar] [CrossRef]
- Gopal, R.; Mendy, A.; Marinelli, M.A.; Richwalls, L.J.; Seger, P.J.; Patel, S.; McHugh, K.J.; Rich, H.E.; Grousd, J.A.; Forno, E.; et al. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance during Influenza-Bacterial Super-Infection. Viruses 2019, 11, 505. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, R.; Gorrochotegui-Escalante, N.; Taddeo, J.; Buttaro, B.; Beld, J.; Tam, V. Eicosanoid-Activated PPARα Inhibits NFκB-Dependent Bacterial Clearance During Post-Influenza Superinfection. Front. Cell. Infect. Microbiol. 2022, 12, 881462. [Google Scholar] [CrossRef]
- Cevallos, S.A.; Lee, J.Y.; Velazquez, E.M.; Foegeding, N.J.; Shelton, C.D.; Tiffany, C.R.; Parry, B.H.; Stull-Lane, A.R.; Olsan, E.E.; Savage, H.P.; et al. 5-Aminosalicylic Acid Ameliorates Colitis and Checks Dysbiotic Escherichia coli Expansion by Activating PPAR-γ Signaling in the Intestinal Epithelium. mBio 2021, 12. [Google Scholar] [CrossRef]
- Ramirez-Moral, I.; Ferreira, B.L.; de Vos, A.F.; van der Poll, T. Post-treatment with the PPAR-γ agonist pioglitazone inhibits inflammation and bacterial growth during Klebsiella pneumonia. Respir. Res. 2021, 22, 230. [Google Scholar] [CrossRef]
- Pu, Q.; Guo, K.; Lin, P.; Wang, Z.; Qin, S.; Gao, P.; Combs, C.; Khan, N.; Xia, Z.; Wu, M. Bitter receptor TAS2R138 facilitates lipid droplet degradation in neutrophils during Pseudomonas aeruginosa infection. Signal Transduct. Target. Ther. 2021, 6, 210. [Google Scholar] [CrossRef]
- Ferreira, B.L.; Ramirez-Moral, I.; Otto, N.A.; Salomão, R.; de Vos, A.F.; van der Poll, T. The PPAR-γ agonist pioglitazone exerts proinflammatory effects in bronchial epithelial cells during acute Pseudomonas aeruginosa pneumonia. Clin. Exp. Immunol. 2022, 207, 370–377. [Google Scholar] [CrossRef]
- Wu, X.; Cheng, B.; Guo, X.; Wu, Q.; Sun, S.; He, P. PPARα/γ signaling pathways are involved in Chlamydia pneumoniae-induced foam cell formation via upregulation of SR-A1 and ACAT1 and downregulation of ABCA1/G1. Microb. Pathog. 2021, 161, 105284. [Google Scholar] [CrossRef]
- Katkar, G.D.; Sayed, I.M.; Anandachar, M.S.; Castillo, V.; Vidales, E.; Toobian, D.; Usmani, F.; Sawires, J.R.; Leriche, G.; Yang, J.; et al. Artificial intelligence-rationalized balanced PPARα/γ dual agonism resets dysregulated macrophage processes in inflammatory bowel disease. Commun. Biol. 2022, 5, 231. [Google Scholar] [CrossRef]
- Wani, K.A.; Goswamy, D.; Taubert, S.; Ratnappan, R.; Ghazi, A.; Irazoqui, J.E. NHR-49/PPAR-α and HLH-30/TFEB cooperate for C. elegans host defense via a flavin-containing monooxygenase. eLife 2021, 10, e62775. [Google Scholar] [CrossRef]
- Liang, F.; Guan, H.; Li, W.; Zhang, X.; Liu, T.; Liu, Y.; Mei, J.; Jiang, C.; Zhang, F.; Luo, B.; et al. Erythropoietin Promotes Infection Resolution and Lowers Antibiotic Requirements in E. coli- and S. aureus-Initiated Infections. Front. Immunol. 2021, 12, 658715. [Google Scholar] [CrossRef]
- Allen, P.E.; Noland, R.C.; Martinez, J.J. Rickettsia conorii survival in THP-1 macrophages involves host lipid droplet alterations and active rickettsial protein production. Cell. Microbiol. 2021, 23, e13390. [Google Scholar] [CrossRef]
- Tanigawa, K.; Luo, Y.; Kawashima, A.; Kiriya, M.; Nakamura, Y.; Karasawa, K.; Suzuki, K. Essential Roles of PPARs in Lipid Metabolism during Mycobacterial Infection. Int. J. Mol. Sci. 2021, 22, 7597. [Google Scholar] [CrossRef]
- Luo, Y.; Tanigawa, K.; Kawashima, A.; Ishido, Y.; Ishii, N.; Suzuki, K. The function of peroxisome proliferator-activated receptors PPAR-γ and PPAR-δ in Mycobacterium leprae-induced foam cell formation in host macrophages. PLoS Negl. Trop. Dis. 2020, 14, e0008850. [Google Scholar] [CrossRef]
- Rajaram, M.V.; Brooks, M.N.; Morris, J.D.; Torrelles, J.B.; Azad, A.K.; Schlesinger, L.S. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J. Immunol. 2010, 185, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.; Dkhar, H.K.; Chandra, V.; Dave, S.; Nanduri, R.; Janmeja, A.K.; Agrewala, J.N.; Gupta, P. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J. Immunol. 2012, 188, 5593–5603. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.E.; Silva, A.R.; Maya-Monteiro, C.M.; Töröcsik, D.; D’Avila, H.; Dezsö, B.; Magalhães, K.G.; Castro-Faria-Neto, H.C.; Nagy, L.; Bozza, P.T. Mycobacterium bovis bacillus Calmette-Guérin infection induces TLR2-dependent peroxisome proliferator-activated receptor gamma expression and activation: Functions in inflammation, lipid metabolism, and pathogenesis. J. Immunol. 2009, 183, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- D’Avila, H.; Melo, R.C.; Parreira, G.G.; Werneck-Barroso, E.; Castro-Faria-Neto, H.C.; Bozza, P.T. Mycobacterium bovis bacillus Calmette-Guérin induces TLR2-mediated formation of lipid bodies: Intracellular domains for eicosanoid synthesis in vivo. J. Immunol. 2006, 176, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-α Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Kim, J.K.; Hanh, B.T.B.; Kim, S.Y.; Kim, H.J.; Kim, Y.J.; Jeon, S.M.; Park, C.R.; Oh, G.T.; Park, J.W.; et al. The Peroxisome Proliferator-Activated Receptor α-Agonist Gemfibrozil Promotes Defense Against Mycobacterium abscessus Infections. Cells 2020, 9, 648. [Google Scholar] [CrossRef] [Green Version]
- Yamanishi, Y.; Miyake, K.; Iki, M.; Tsutsui, H.; Karasuyama, H. Recent advances in understanding basophil-mediated Th2 immune responses. Immunol. Rev. 2017, 278, 237–245. [Google Scholar] [CrossRef]
- Chen, T.; Tibbitt, C.A.; Feng, X.; Stark, J.M.; Rohrbeck, L.; Rausch, L.; Sedimbi, S.K.; Karlsson, M.C.I.; Lambrecht, B.N.; Karlsson Hedestam, G.B.; et al. PPAR-γ promotes type 2 immune responses in allergy and nematode infection. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- He, X.; Gong, P.; Wei, Z.; Liu, W.; Wang, W.; Li, J.; Yang, Z.; Zhang, X. Peroxisome proliferator-activated receptor-γ-mediated polarization of macrophages in Neospora caninum infection. Exp. Parasitol. 2017, 178, 37–44. [Google Scholar] [CrossRef]
- Chen, K.M.; Peng, C.Y.; Shyu, L.Y.; Lan, K.P.; Lai, S.C. Peroxisome-Proliferator Activator Receptor γ in Mouse Model with Meningoencephalitis Caused by Angiostrongylus cantonensis. J. Parasitol. 2021, 107, 205–213. [Google Scholar] [CrossRef]
- Mita-Mendoza, N.K.; Magallon-Tejada, A.; Parmar, P.; Furtado, R.; Aldrich, M.; Saidi, A.; Taylor, T.; Smith, J.; Seydel, K.; Daily, J.P. Dimethyl fumarate reduces TNF and Plasmodium falciparum induced brain endothelium activation in vitro. Malar. J. 2020, 19, 376. [Google Scholar] [CrossRef]
- Borges, T.K.; Alves, É.A.; Vasconcelos, H.A.; Carneiro, F.P.; Nicola, A.M.; Magalhães, K.G.; Muniz-Junqueira, M.I. Differences in the modulation of reactive species, lipid bodies, cyclooxygenase-2, 5-lipoxygenase and PPAR-γ in cerebral malaria-susceptible and resistant mice. Immunobiology 2017, 222, 604–619. [Google Scholar] [CrossRef]
- Matta, S.K.; Rinkenberger, N.; Dunay, I.R.; Sibley, L.D. Toxoplasma gondii infection and its implications within the central nervous system. Nat. Rev. Microbiol. 2021, 19, 467–480. [Google Scholar] [CrossRef]
- Shyu, L.Y.; Chen, K.M.; Lu, C.Y.; Lai, S.C. Regulation of Proinflammatory Enzymes by Peroxisome Proliferator-Activated Receptor Gamma in Astroglia Infected with Toxoplasma gondii. J. Parasitol. 2020, 106, 564–571. [Google Scholar] [CrossRef]
- He, J.J.; Ma, J.; Elsheikha, H.M.; Song, H.Q.; Zhou, D.H.; Zhu, X.Q. Proteomic Profiling of Mouse Liver following Acute Toxoplasma gondii Infection. PLoS ONE 2016, 11, e0152022. [Google Scholar] [CrossRef] [Green Version]
- Moulik, S.; Karmakar, J.; Joshi, S.; Dube, A.; Mandal, C.; Chatterjee, M. Status of IL-4 and IL-10 driven markers in experimental models of Visceral Leishmaniasis. Parasite Immunol. 2021, 43, e12783. [Google Scholar] [CrossRef]
- McManus, D.P.; Dunne, D.W.; Sacko, M.; Utzinger, J.; Vennervald, B.J.; Zhou, X.N. Schistosomiasis. Nat. Rev. Dis. Prim. 2018, 4, 13. [Google Scholar] [CrossRef]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef]
- Shiels, J.; Cwiklinski, K.; Alvarado, R.; Thivierge, K.; Cotton, S.; Gonzales Santana, B.; To, J.; Donnelly, S.; Taggart, C.C.; Weldon, S.; et al. Schistosoma mansoni immunomodulatory molecule Sm16/SPO-1/SmSLP is a member of the trematode-specific helminth defence molecules (HDMs). PLoS Negl. Trop. Dis. 2020, 14, e0008470. [Google Scholar] [CrossRef]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Penas, F.; Mirkin, G.A.; Vera, M.; Cevey, Á.; González, C.D.; Gómez, M.I.; Sales, M.E.; Goren, N.B. Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 2015, 1852, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Penas, F.N.; Carta, D.; Cevey, Á.C.; Rada, M.J.; Pieralisi, A.V.; Ferlin, M.G.; Sales, M.E.; Mirkin, G.A.; Goren, N.B. Pyridinecarboxylic Acid Derivative Stimulates Pro-Angiogenic Mediators by PI3K/AKT/mTOR and Inhibits Reactive Nitrogen and Oxygen Species and NF-κB Activation Through a PPARγ-Dependent Pathway in T. cruzi-Infected Macrophages. Front. Immunol. 2019, 10, 2955. [Google Scholar] [CrossRef]
- Penas, F.N.; Cevey, Á.C.; Siffo, S.; Mirkin, G.A.; Goren, N.B. Hepatic injury associated with Trypanosoma cruzi infection is attenuated by treatment with 15-deoxy-Δ(12,14) prostaglandin J(2). Exp. Parasitol. 2016, 170, 100–108. [Google Scholar] [CrossRef]
- González, F.B.; Villar, S.R.; Toneatto, J.; Pacini, M.F.; Márquez, J.; D’Attilio, L.; Bottasso, O.A.; Piwien-Pilipuk, G.; Pérez, A.R. Immune response triggered by Trypanosoma cruzi infection strikes adipose tissue homeostasis altering lipid storage, enzyme profile and adipokine expression. Med. Microbiol. Immunol. 2019, 208, 651–666. [Google Scholar] [CrossRef]
- Dreesen, L.; De Bosscher, K.; Grit, G.; Staels, B.; Lubberts, E.; Bauge, E.; Geldhof, P. Giardia muris infection in mice is associated with a protective interleukin 17A response and induction of peroxisome proliferator-activated receptor alpha. Infect. Immun. 2014, 82, 3333–3340. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Pathogen | Study Model | Intervention | PPAR Status | Mechanism | Ref. |
|---|---|---|---|---|---|
| IAV, RSV | AMs, mice | PpargΔLyz2 mice | ↓ | Regulation of PPARγ through STAT1 activation following IFN signaling | [76] |
| IAV | AMs, human lung macrophages, mice | PpargΔLyz2 mice, Bleomycin | ↓ | Increased influenza-induced pulmonary collagen deposition in PPARγ-deficient mice | [78] |
| IAV | A549 cells, mice | Emodin and its analogs | ↑ | Activation of PPARα/γ and AMPK, decreased fatty acid biosynthesis and increased ATP level | [79] |
| MERS-CoV | THP-1 cells, primary human monocytes | siRNAs | ↑ | MERS-CoV S glycoprotein interaction with DPP4 leading to IRAK-M and PPARγ expression | [87] |
| CHIKV | Vero cells, RAW264.7 cells | Telmisatran, PPAR-γ antagonist GW9662 | ↓ | Activation of PPAR-γ and inhibition of AT1 by telmisartan | [88] |
| HIV | Primary rat astrocytes, microglia, rats | gp120ADA, Rosiglitazone, Pioglitazone | ↓ | Induction of inflammatory response and decrease in GLT-1 expression in the brain by gp120 | [89] |
| HBV | HepG2.2.15, Huh7, HepG2-NTCP ells | OS_128167, overexpression and downregulation studies, HBV transgenic mice | - | Activation of HBV core promoter by SIRT6 through upregulation of PPARα | [90] |
| HCV | Huh7.5 cells | Calciterol, Linoleic acid, Ly171883, Wy14643 | - | Activation of VDR but inhibition of PPARα/β/γ by calcitriol | [91] |
| Pathogen | Drug/Reagent | Function | Study Model | Mechanism of Action | Ref. |
|---|---|---|---|---|---|
| Escherichia coli | 5-aminosalicylic acid | PPARγ agonist | DSS-induced murine colitis model, Pparg-deficient mice, CaCo-2 cells | Amelioration of a respiration-dependent luminal expansion of E. coli | [104] |
| Klebsiella pneumoniae | Pioglitazone | PPARγ agonist | In vivo mouse model | Reduction of cytokines and myeloperoxidase levels in the lungs | [105] |
| Pseudomonas aeruginosa | Pioglitazone | PPARγ agonist | In vivo mouse model | Increased pro-inflammatory cytokines with enhanced expression of genes involved in glycolysis | [107] |
| Chlamydia pneumoniae | Rosiglitazone | PPARγ agonist | THP-1 macrophages, HEp-2 cells | Regulation of Cpn induced macrophage-derived foam cell formation by upregulating SR-A1 an ACAT1, and downregulating ABCA1/G1 expression via PPARα/γ signaling | [108] |
| Fenofibrate | PPARα agonist | ||||
| GW9662 | PPARγ antagonist | ||||
| MK886 | PPARα antagonist | ||||
| Citrobacter rodentium | PAR5359 | PPARα/γ-dual-agonist | Citrobacter rodentium- and DSS-induced murine colitis model, IBD patient-derived PBMCs | Enhanced bacterial clearance, controlled production of ROS and cytokines, anti-inflammatory/healing | [109] |
| Rickettsia conorii | GW9662 | PPARγ antagonist | THP-1 macrophages | Increased intracellular survival of bacteria | [112] |
| Pathogen | Drug/Reagent | Function | Study Model | Mechanism of Action | Ref. |
|---|---|---|---|---|---|
| Angiostrongylus cantonensis | GW9662 | PPARγ antagonist | Mouse model of angiostrongyliasis | NF-κB activation and increase in inflammation and BBB permeability | [124] |
| Plasmodium falciparum | Dimethyl fumarate | - | Cerebral cortex derived HBMVECs | Upregulation of PPAR pathway, NRF2-mediated oxidative stress responses, ErbB4 signaling to downregulate the neuroinflammation | [125] |
| Toxoplasma gondii | Rosiglitazone | PPARγ agonist | SVG p12 cells, Hs68 cells | Decreased expression of MMP-2, MMP-9, COX-2, PGE2, iNOS and NO | [128] |
| GW9662 | PPARγ antagonist | Increased expression of MMP-2, MMP-9, COX-2, PGE2, iNOS and NO | |||
| Trypanosoma cruzi | HP24 | pyridinecarboxylic acid derivative | In vivo mice infection, mouse peritoneal macrophages | Induction of PI3K/Akt/mTOR signaling (pro-angiogenic), inhibition of NF-κB signaling (anti-inflammatory) | [136] |
| 15-deoxy-D12,14 prostaglandin J2 | PPARγ agonist | In vivo mice infection | Reduction of liver inflammatory infiltrates, pro-inflammatory enzymes and cytokine expression through inhibition of NF-kB signaling, No change in parasitic load | [137] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, I.S.; Silwal, P.; Jo, E.-K. Peroxisome Proliferator-Activated Receptor-Targeted Therapies: Challenges upon Infectious Diseases. Cells 2023, 12, 650. https://doi.org/10.3390/cells12040650
Kim IS, Silwal P, Jo E-K. Peroxisome Proliferator-Activated Receptor-Targeted Therapies: Challenges upon Infectious Diseases. Cells. 2023; 12(4):650. https://doi.org/10.3390/cells12040650
Chicago/Turabian StyleKim, In Soo, Prashanta Silwal, and Eun-Kyeong Jo. 2023. "Peroxisome Proliferator-Activated Receptor-Targeted Therapies: Challenges upon Infectious Diseases" Cells 12, no. 4: 650. https://doi.org/10.3390/cells12040650
APA StyleKim, I. S., Silwal, P., & Jo, E. -K. (2023). Peroxisome Proliferator-Activated Receptor-Targeted Therapies: Challenges upon Infectious Diseases. Cells, 12(4), 650. https://doi.org/10.3390/cells12040650