


Influence of Immune System Abnormalities Caused by Maternal Immune Activation in the Postnatal Period


Abstract
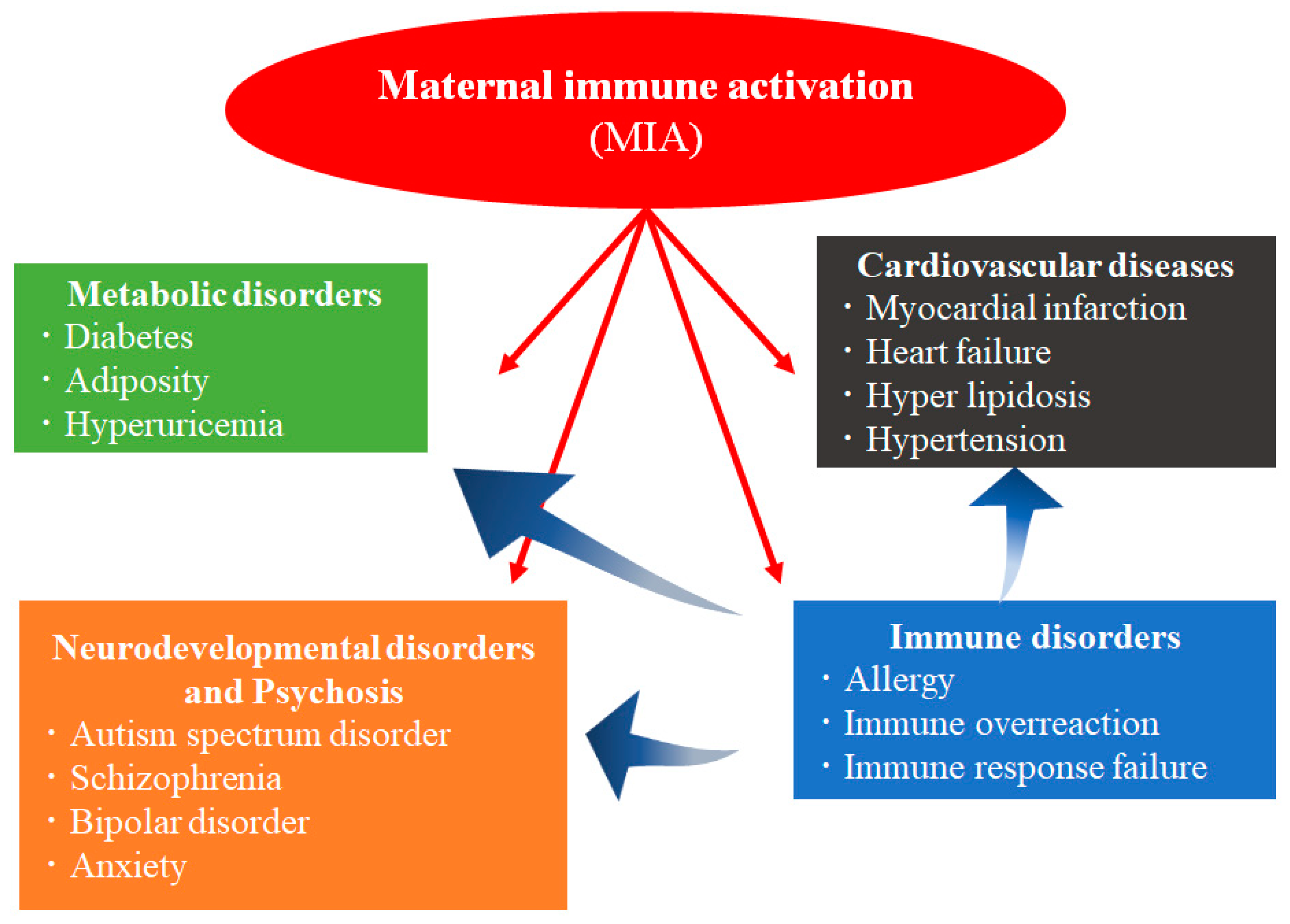
:1. Introduction
2. Immunological Disorders Caused by MIA in Humans
2.1. Diseases Associated with an Abnormal Immunity Induced by Maternal Infection in Humans
2.2. Mechanism of Diseases and Disorders with Abnormal Immunity Caused by Maternal Infection
2.2.1. Diabetes Mellitus
2.2.2. Allergic Diseases
2.2.3. Neurodevelopmental Disorders and Psychosis
3. The Impact of Exposure to SARS-CoV-2 Infections in the Prenatal and Postnatal Period
3.1. Possible Relationship between Prenatal Environment and COVID-19 Infection in the Postnatal Period
3.2. Adverse Effects in Infants Exposed to COVID-19 Infection in the Prenatal Period
3.2.1. Neurodevelopmental Disorders
3.2.2. Immune Dysfunction
3.2.3. Endothelial Cell Dysfunction
4. Immune Dysfunction Caused by MIA in Animal Models
4.1. Immune Dysfunction Caused by Prenatal Exposure to Poly (I:C)
4.2. Immune Dysfunction Caused by Prenatal Exposure to LPS
5. The Gestation Period, Inflammatory Magnitude, Inflammatory Type of MIA, and Immune Dysfunction Mechanism in Offspring
5.1. Alteration of the Immune System Affected by the Time of MIA
5.2. Alteration of the Immune System Due to the Magnitude of Inflammatory Response or Type of Inflammatory Cytokines Present in the Prenatal Period
6. Epigenetic Changes in the Immune System
6.1. Importance of Epigenetic Alterations in Life
6.2. Epigenetic Changes Induced by MIA in the Immune System
6.3. Prospects of Prevention and Treatment Using Epigenetic Therapy in the Prenatal Period
7. Conclusions
The Association of Genetic Factors, Maternal Infection, and Postnatal Environmental Factors in Immune Dysfunction
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, T.; Liu, H.-X.; Yan, H.-Y.; Wu, D.-M.; Ping, J. Developmental origins of inflammatory and immune diseases. Mol. Hum. Reprod. 2016, 22, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 1, 1077–1081. [Google Scholar] [CrossRef]
- Barker, D.J.; Winter, P.D.; Osmond, C.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 2, 577–580. [Google Scholar] [CrossRef]
- Morea, M.; Miu, N.; Morea, V.F.; Cornean, R. Maternal obesity—A risk factor for metabolic syndrome in children. Clujul Med 2013, 86, 259–265. [Google Scholar]
- Nilsen, F.M.; Ruiz, J.D.; Tulve, N.S. A Meta-Analysis of Stressors from the Total Environment Associated with Children’s General Cognitive Ability. Int. J. Environ. Res. Public Health 2020, 17, 5451. [Google Scholar] [CrossRef]
- Weijmans, M.; van der Graaf, Y.; Reitsma, J.; Visseren, F. Paternal or maternal history of cardiovascular disease and the risk of cardiovascular disease in offspring. A systematic review and meta-analysis. Int. J. Cardiol. 2015, 179, 409–416. [Google Scholar] [CrossRef]
- Fuchs, O.; von Mutius, E. Prenatal and childhood infections: Implications for the development and treatment of childhood asthma. Lancet Respir. Med. 2013, 1, 743–754. [Google Scholar] [CrossRef]
- Burke, H.; Leonardi-Bee, J.; Hashim, A.; Pine-Abata, H.; Chen, Y.; Cook, D.G.; Britton, J.R.; McKeever, T.M. Prenatal and Passive Smoke Exposure and Incidence of Asthma and Wheeze: Systematic Review and Meta-analysis. Pediatrics 2012, 129, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Prenatal risk factors for autism: Comprehensive meta-analysis. Br. J. Psychiatry 2009, 195, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.N.; Van de Water, J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J. Neuroimmunol. 2011, 232, 196–199. [Google Scholar] [CrossRef] [Green Version]
- Terasaki, L.S.; Schwarz, J.M. Effects of Moderate Prenatal Alcohol Exposure during Early Gestation in Rats on Inflammation across the Maternal-Fetal-Immune Interface and Later-Life Immune Function in the Offspring. J. Neuroimmune Pharmacol. 2016, 11, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Shcherbitskaia, A.D.; Vasilev, D.S.; Milyutina, Y.P.; Tumanova, N.L.; Zalozniaia, I.V.; Kerkeshko, G.O.; Arutjunyan, A.V. Maternal Hyperhomocysteinemia Induces Neuroinflammation and Neuronal Death in the Rat Offspring Cortex. Neurotox. Res. 2020, 38, 408–420. [Google Scholar] [CrossRef]
- Jain, S.; Baer, R.J.; McCulloch, C.E.; Rogers, E.; Rand, L.; Jelliffe-Pawlowski, L.; Piao, X. Association of Maternal Immune Activation during Pregnancy and Neurologic Outcomes in Offspring. J. Pediatr. 2021, 238, 87–93.e3. [Google Scholar] [CrossRef]
- McKeever, T.M.; Lewis, S.A.; Smith, C.; Hubbard, R. The importance of prenatal exposures on the development of allergic disease: A birth cohort study using the West Midlands General Practice Database. Am. J. Respir. Crit. Care Med. 2002, 166, 827–832. [Google Scholar] [CrossRef]
- Yue, Y.; Tang, Y.; Tang, J.; Shi, J.; Zhu, T.; Huang, J.; Qiu, X.; Zeng, Y.; Li, W.; Qu, Y.; et al. Maternal infection during pregnancy and type 1 diabetes mellitus in offspring: A systematic review and meta-analysis. Epidemiology Infect. 2018, 146, 2131–2138. [Google Scholar] [CrossRef] [Green Version]
- Enstrom, A.M.; Onore, C.E.; Van de Water, J.A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain, Behav. Immun. 2010, 24, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain, Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Antoun, S.; Ellul, P.; Peyre, H.; Rosenzwajg, M.; Gressens, P.; Klatzmann, D.; Delorme, R. Fever during pregnancy as a risk factor for neurodevelopmental disorders: Results from a systematic review and meta-analysis. Mol. Autism 2021, 12, 60. [Google Scholar] [CrossRef]
- Han, V.X.; Patel, S.; Jones, H.F.; Nielsen, T.C.; Mohammad, S.S.; Hofer, M.J.; Gold, W.; Brilot, F.; Lain, S.J.; Nassar, N.; et al. Maternal acute and chronic inflammation in pregnancy is associated with common neurodevelopmental disorders: A systematic review. Transl. Psychiatry 2021, 11, 71. [Google Scholar] [CrossRef]
- Zinkernagel, R.M. Maternal antibodies, childhood infections, and autoimmune diseases. N. Engl. J. Med. 2001, 345, 1331–1335. [Google Scholar] [CrossRef]
- Hermann, E.; Truyens, C.; Alonso-Vega, C.; Even, J.; Rodriguez, P.; Berthe, A.; Gonzalez-Merino, E.; Torrico, F.; Carlier, Y. Human fetuses are able to mount an adultlike CD8 T-cell response. Blood 2002, 100, 2153–2158. [Google Scholar] [CrossRef] [PubMed]
- Douwes, J.; Cheng, S.; Travier, N.; Cohet, C.; Niesink, A.; McKenzie, J.; Cunningham, C.; Le Gros, G.; von Mutius, E.; Pearce, N. Farm exposure in utero may protect against asthma, hay fever and eczema. Eur. Respir. J. 2008, 32, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Liu, X.; Li, Y.; Meng, S.; Wu, F.; Yan, B.; Xue, Y.; Ma, T.; Yang, J.; Liu, J. Maternal exposure to farming environment protects offspring against allergic diseases by modulating the neonatal TLR-Tregs-Th axis. Clin. Transl. Allergy 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Wan, C.-L.; He, L.; Jong, S.; Chou, K.-C. Gestational Influenza Increases the Risk of Psychosis in Adults. Med. Chem. 2015, 11, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Quagliato, L.A.; de Matos, U.; Nardi, A.E. Maternal immune activation generates anxiety in offspring: A translational meta-analysis. Transl. Psychiatry 2021, 11, 245. [Google Scholar] [CrossRef]
- Oncu-Oner, T.; Can, S. Meta-analysis of the relationship between Toxoplasma gondii and schizophrenia. Ann. Parasitol. 2022, 68, 103–110. [Google Scholar]
- Davies, C.; Segre, G.; Estradé, A.; Radua, J.; De Micheli, A.; Provenzani, U.; Oliver, D.; de Pablo, G.S.; Ramella-Cravaro, V.; Besozzi, M.; et al. Prenatal and perinatal risk and protective factors for psychosis: A systematic review and meta-analysis. Lancet Psychiatry 2020, 7, 399–410. [Google Scholar] [CrossRef]
- Selten, J.-P.; Termorshuizen, F. The serological evidence for maternal influenza as risk factor for psychosis in offspring is insufficient: Critical review and meta-analysis. Schizophr. Res. 2017, 183, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-Y.; Xu, L.-L.; Shao, L.; Xia, R.-M.; Yu, Z.-H.; Ling, Z.-X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain, Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef]
- Hu, C.-C.; Xu, X.; Xiong, G.-L.; Xu, Q.; Zhou, B.-R.; Li, C.-Y.; Qin, Q.; Liu, C.-X.; Li, H.-P.; Sun, Y.-J.; et al. Alterations in plasma cytokine levels in chinese children with autism spectrum disorder. Autism Res. 2018, 11, 989–999. [Google Scholar] [CrossRef]
- Eftekharian, M.M.; Ghafouri-Fard, S.; Noroozi, R.; Omrani, M.D.; Arsang-Jang, S.; Ganji, M.; Gharzi, V.; Noroozi, H.; Komaki, A.; Mazdeh, M.; et al. Cytokine profile in autistic patients. Cytokine 2018, 108, 120–126. [Google Scholar] [CrossRef]
- Molloy, C.; Morrow, A.; Meinzenderr, J.; Schleifer, K.; Dienger, K.; Manningcourtney, P.; Altaye, M.; Willskarp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef]
- Guloksuz, S.A.; Abali, O.; Aktas Cetin, E.; Bilgic Gazioglu, S.; Deniz, G.; Yildirim, A.; Kawikova, I.; Guloksuz, S.; Leckman, J.F. Elevated plasma concentrations of S100 calcium-binding protein B and tumor necrosis factor alpha in children with autism spectrum disorders. Braz. J. Psychiatry 2017, 39, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Huang, L.; Li, X.; Li, H.; Zhou, Y.; Zhu, H.; Pan, T.; Kendrick, K.M.; Xu, W. Immunological cytokine profiling identifies TNF-α as a key molecule dysregulated in autistic children. Oncotarget 2017, 8, 82390–82398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jácome, M.C.I.; Chacòn, L.M.M.; Cuesta, H.V.; Rizo, C.M.; Santiesteban, M.W.; Hernandez, L.R.; García, E.N.; Fraguela, M.E.G.; Verdecia, C.I.F.; Hurtado, Y.V.; et al. Peripheral Inflammatory Markers Contributing to Comorbidities in Autism. Behav. Sci. 2016, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Matsuzaki, H.; Iwata, K.; Kameno, Y.; Shimmura, C.; Kawai, S.; Yoshihara, Y.; Wakuda, T.; Takebayashi, K.; Takagai, S.; et al. Plasma Cytokine Profiles in Subjects with High-Functioning Autism Spectrum Disorders. PLoS ONE 2011, 6, e20470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ayadhi, L.Y. Pro-inflammatory cytokines in autistic children in central Saudi Arabia. Neurosciences 2005, 10, 155–158. [Google Scholar] [PubMed]
- Han, V.X.; Jones, H.F.; Patel, S.; Mohammad, S.S.; Hofer, M.J.; Alshammery, S.; Maple-Brown, E.; Gold, W.; Brilot, F.; Dale, R.C. Emerging evidence of Toll-like receptors as a putative pathway linking maternal inflammation and neurodevelopmental disorders in human offspring: A systematic review. Brain Behav. Immun. 2022, 99, 91–105. [Google Scholar] [CrossRef]
- Bundo, M.; Toyoshima, M.; Okada, Y.; Akamatsu, W.; Ueda, J.; Nemoto-Miyauchi, T.; Sunaga, F.; Toritsuka, M.; Ikawa, D.; Kakita, A.; et al. Increased L1 Retrotransposition in the Neuronal Genome in Schizophrenia. Neuron 2014, 81, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Chen, Y.; Zhao, D.; Liu, T.; Huang, Y.; Qiu, L.; Hao, Y.; Hu, X.; Yin, W.; Liu, Z.; et al. Recommendations for the Diagnosis, Prevention, and Control of Coronavirus Disease-19 in Children-The Chinese Perspectives. Front. Pediatr. 2020, 8, 553394. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, Y. The Clinical Characteristics and Risk Factors of Severe COVID-19. Gerontology 2021, 67, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.S.; Sehayek, D.; Gabrielli, S.; Zhang, X.; McCusker, C.; Ben-Shoshan, M. COVID-19 and comorbidities: A systematic review and meta-analysis. Postgrad. Med. 2020, 132, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Parohan, M.; Yaghoubi, S.; Seraji, A.; Javanbakht, M.H.; Sarraf, P.; Djalali, M. Risk factors for mortality in patients with Coronavirus disease 2019 (COVID-19) infection: A systematic review and meta-analysis of observational studies. Aging Male 2020, 23, 1416–1424. [Google Scholar] [CrossRef]
- Zheng, Z.; Peng, F.; Xu, B.; Zhao, J.; Liu, H.; Peng, J.; Li, Q.; Jiang, C.; Zhou, Y.; Liu, S.; et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 2020, 81, e16–e25. [Google Scholar] [CrossRef]
- Yildirim, Z.; Sahin, O.S.; Yazar, S.; Cetintas, V.B. Genetic and epigenetic factors associated with increased severity of Covid-19. Cell Biol. Int. 2021, 45, 1158–1174. [Google Scholar] [CrossRef]
- Castro de Moura, M.; Davalos, V.; Planas-Serra, L.; Alvarez-Errico, D.; Arribas, C.; Ruiz, M.; Aguilera-Albesa, S.; Troya, J.; Valencia-Ramos, J.; Vélez-Santamaria, V.; et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. eBioMedicine 2021, 66, 103339. [Google Scholar] [CrossRef]
- Edlow, A.G.; Castro, V.M.; Shook, L.L.; Kaimal, A.J.; Perlis, R.H. Neurodevelopmental Outcomes at 1 Year in Infants of Mothers Who Tested Positive for SARS-CoV-2 During Pregnancy. JAMA Netw. Open 2022, 5, e2215787. [Google Scholar] [CrossRef]
- Manti, S.; Leonardi, S.; Rezaee, F.; Harford, T.J.; Perez, M.K.; Piedimonte, G. Effects of Vertical Transmission of Respiratory Viruses to the Offspring. Front. Immunol. 2022, 13, 853009. [Google Scholar] [CrossRef]
- Gee, S.; Chandiramani, M.; Seow, J.; Pollock, E.; Modestini, C.; Das, A.; Tree, T.; Doores, K.J.; Tribe, R.M.; Gibbons, D.L. The legacy of maternal SARS-CoV-2 infection on the immunology of the neonate. Nat. Immunol. 2021, 22, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Liu, F.; Blair, R.; Wang, C.; Yang, H.; Mudd, J.; Currey, J.M.; Iwanaga, N.; He, J.; Mi, R.; et al. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics 2021, 11, 8076–8091. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-W.; Ilyas, I.; Weng, J.-P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2022, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sharps, M.C.; Hayes, D.J.; Lee, S.; Zou, Z.; Brady, C.A.; Almoghrabi, Y.; Kerby, A.; Tamber, K.K.; Jones, C.J.; Waldorf, K.M.A.; et al. A structured review of placental morphology and histopathological lesions associated with SARS-CoV-2 infection. Placenta 2020, 101, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, B.; Barratt, I.; Townsend, R.; Kalafat, E.; van der Meulen, J.; Gurol-Urganci, I.; O’Brien, P.; Morris, E.; Draycott, T.; Thangaratinam, S.; et al. Effects of the COVID-19 pandemic on maternal and perinatal outcomes: A systematic review and meta-analysis. Lancet Glob. Health 2021, 9, e759–e772. [Google Scholar] [CrossRef]
- Wei, S.Q.; Bilodeau-Bertrand, M.; Liu, S.; Auger, N. The impact of COVID-19 on pregnancy outcomes: A systematic review and meta-analysis. Can. Med Assoc. J. 2021, 193, E540–E548. [Google Scholar] [CrossRef]
- Lassi, Z.S.; Ana, A.; Das, J.K.; A Salam, R.; A Padhani, Z.; Irfan, O.; A Bhutta, Z. A systematic review and meta-analysis of data on pregnant women with confirmed COVID-19: Clinical presentation, and pregnancy and perinatal outcomes based on COVID-19 severity. J. Glob. Health 2021, 11, 05018. [Google Scholar] [CrossRef]
- Shimizu, Y.; Tsukada, T.; Sakata-Haga, H.; Sakai, D.; Shoji, H.; Saikawa, Y.; Hatta, T. Exposure to Maternal Immune Activation Causes Congenital Unfolded Protein Response Defects and Increases the Susceptibility to Postnatal Inflammatory Stimulation in Offspring. J. Inflamm. Res. 2021, 14, 355–365. [Google Scholar] [CrossRef]
- Rose, D.R.; Careaga, M.; Van de Water, J.; McAllister, K.; Bauman, M.D.; Ashwood, P. Long-term altered immune responses following fetal priming in a non-human primate model of maternal immune activation. Brain, Behav. Immun. 2017, 63, 60–70. [Google Scholar] [CrossRef]
- Onore, C.E.; Schwartzer, J.J.; Careaga, M.; Berman, R.F.; Ashwood, P. Maternal immune activation leads to activated inflammatory macrophages in offspring. Brain, Behav. Immun. 2014, 38, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Hameete, B.C.; Fernández-Calleja, J.M.; de Groot, M.W.; Oppewal, T.R.; Tiemessen, M.M.; Hogenkamp, A.; de Vries, R.B.; Groenink, L. The poly(I:C)-induced maternal immune activation model; a systematic review and meta-analysis of cytokine levels in the offspring. Brain, Behav. Immun. Health 2021, 11, 100192. [Google Scholar] [CrossRef]
- Tartaglione, A.M.; Villani, A.; Ajmone-Cat, M.A.; Minghetti, L.; Ricceri, L.; Pazienza, V.; De Simone, R.; Calamandrei, G. Maternal immune activation induces autism-like changes in behavior, neuroinflammatory profile and gut microbiota in mouse offspring of both sexes. Transl. Psychiatry 2022, 12, 1–10. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Yim, Y.S.; Ha, S.; Atarashi, K.; Tan, T.G.; Longman, R.S.; Honda, K.; Littman, D.R.; Choi, G.B.; et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 2017, 549, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, E.Y.; McBride, S.W.; Chow, J.; Mazmanian, S.K.; Patterson, P.H. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 12776–12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Committee on Infectious Diseases. Recommendations for Prevention and Control of Influenza in Children, 2018–2019. Pediatrics 2018, 142, e20182367. [Google Scholar] [CrossRef] [PubMed]
- Ter Haar, N.; Oswald, M.; Jeyaratnam, J.; Anton, J.; Barron, K.; Brogan, P.; Cantarini, L.; Galeotti, C.; Grateau, G.; Hentgen, V.; et al. Recommendations for the management of autoinflammatory diseases. Pediatr. Rheumatol. 2015, 13, P133. [Google Scholar] [CrossRef] [Green Version]
- Hidaka, F.; Matsuo, S.; Muta, T.; Takeshige, K.; Mizukami, T.; Nunoi, H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin. Immunol. 2006, 119, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Morichi, S.; Okumara, A.; Nakagawa, S.; Morishima, T.; the collaborating study group on influenza-associated encephalopathy in Japan. National survey of pandemic influenza A (H1N1) 2009-associated encephalopathy in Japanese children. J. Med Virol. 2012, 84, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.-I.; Nagashima, M.; Sasaki, Y.; Mori, K.; Tabei, Y.; Yoshida, Y.; Yamazaki, K.; Hirata, I.; Sekine, H.; Ito, T.; et al. A novel amino acid substitution at the receptor-binding site on the hemagglutinin of H3N2 influenza A viruses isolated from 6 cases with acute encephalopathy during the 1997-1998 season in Tokyo. Arch. Virol. 1999, 144, 147–155. [Google Scholar] [CrossRef]
- Surriga, O.; Ortega, A.; Jadeja, V.; Bellafronte, A.; Lasala, N.; Zhou, H. Altered hepatic inflammatory response in the offspring following prenatal LPS exposure. Immunol Lett. 2009, 123, 88–95. [Google Scholar] [CrossRef]
- Hsueh, P.T.; Lin, H.H.; Wang, H.H.; Liu, C.L.; Ni, W.F.; Liu, J.K.; Chang, H.-H.; Sun, D.-S.; Chen, Y.-S.; Chen, Y.-L. Immune imbalance of global gene expression, and cytokine, chemokine and selectin levels in the brains of offspring with social deficits via maternal immune activation. Genes Brain Behav. 2018, 17, e12479. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Lippi, L.L.; Bevilacqua, E.; Bernardi, M.M. LPS exposure increases maternal corticosterone levels, causes placental injury and increases IL-1Β levels in adult rat offspring: Relevance to autism. PLoS ONE 2013, 8, e82244. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Wang, J.; Zhu, Y.; Tseu, I.; Post, M. Maternal endotoxin exposure attenuates allergic airway disease in infant rats. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Basta-Kaim, A.; Szczęsny, E.; Leśkiewicz, M.; Głombik, K.; Ślusarczyk, J.; Budziszewska, B.; Regulska, M.; Kubera, M.; Nowak, W.; Wędzony, K.; et al. Maternal immune activation leads to age-related behavioral and immunological changes in male rat offspring—The effect of antipsychotic drugs. Pharmacol Rep. 2012, 64, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Hodyl, N.A.; Krivanek, K.M.; Lawrence, E.; Clifton, V.L.; Hodgson, D.M. Prenatal exposure to a pro-inflammatory stimulus causes delays in the development of the innate immune response to LPS in the offspring. J. Neuroimmunol. 2007, 190, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, K.; Avagyan, A.; Seib, C.; Frei, R.; Steinle, J.; Ahrens, B.; Dittrich, A.M.; Blumchen, K.; Lauener, R.; Hamelmann, E. Prenatal initiation of endotoxin airway exposure prevents subsequent allergen-induced sensitization and airway inflammation in mice. J. Allergy Clin. Immunol. 2006, 118, 666–673. [Google Scholar] [CrossRef]
- Blümer, N.; Herz, U.; Wegmann, M.; Renz, H. Prenatal lipopolysaccharide-exposure prevents allergic sensitization and airway inflammation, but not airway responsiveness in a murine model of experimental asthma. Clin. Exp. Allergy 2005, 35, 397–402. [Google Scholar] [CrossRef]
- Kirsten, T.B.; De Oliveira, B.P.S.; De Oliveira, A.P.L.; Kieling, K.; De Lima, W.T.; Palermo-Neto, J.; Bernardi, M.M. Single early prenatal lipopolysaccharide exposure prevents subsequent airway inflammation response in an experimental model of asthma. Life Sci. 2011, 89, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.C.M.; Smith, C. Exposure to Maternal Chronic Inflammation Transfers a Pro-Inflammatory Profile to Generation F2 via Sex-Specific Mechanisms. Front Immunol. 2020, 11, 48. [Google Scholar] [CrossRef]
- Lasala, N.; Zhou, H. Effects of maternal exposure to LPS on the inflammatory response in the offspring. J Neuroimmunol. 2007, 189, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.S. Prenatal Infection as a Risk Factor for Schizophrenia. Schizophr. Bull. 2006, 32, 200–202. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. TLR signaling. Semin. Immunol. 2007, 19, 24–32. [Google Scholar] [CrossRef]
- Landreth, K.S. Critical windows in development of the rodent immune system. Hum. Exp. Toxicol. 2002, 21, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Jardine, L.; Gottgens, B.; Teichmann, S.A.; Haniffa, M. Prenatal development of human immunity. Science 2020, 368, 600–603. [Google Scholar] [CrossRef]
- Brown, A.S.; Begg, M.D.; Gravenstein, S.; Schaefer, C.A.; Wyatt, R.J.; Bresnahan, M.; Babulas, V.P.; Susser, E.S. Serologic Evidence of Prenatal Influenza in the Etiology of Schizophrenia. Arch. Gen. Psychiatry 2004, 61, 774–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, D.; St-Amour, I.; Cisbani, G.; Rousseau, L.-S.; Cicchetti, F. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav. Immun. 2014, 38, 77–90. [Google Scholar] [CrossRef]
- Masi, A.; Quintana, D.S.; Glozier, N.; Lloyd, A.R.; Hickie, I.B.; Guastella, A.J. Cytokine aberrations in autism spectrum disorder: A systematic review and meta-analysis. Mol. Psychiatry 2015, 20, 440–446. [Google Scholar] [CrossRef]
- Goines, P.E.; Ashwood, P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicology Teratol. 2013, 36, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.E.P.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal Immune Activation Alters Fetal Brain Development through Interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, Q.; Wang, J.; Tang, M.; Huang, S.; Peng, K.; Han, Y.; Zhang, J.; Liu, G.; Fang, Q.; et al. Maternal immune activation-induced PPARγ-dependent dysfunction of microglia associated with neurogenic impairment and aberrant postnatal behaviors in offspring. Neurobiol. Dis. 2019, 125, 1–13. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J.; Schedlowski, M.; Yee, B.K. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci. Biobehav. Rev. 2005, 29, 913–947. [Google Scholar] [CrossRef]
- Quina, A.; Buschbeck, M.; Di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Power, B.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta (BBA) Rev. Cancer 2007, 1775, 138–162. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Slieker, R.C.; Stein, A.D.; Suchiman, H.E.D.; Slagboom, P.E.; Van Zwet, E.W.; Heijmans, B.T.; Lumey, L.H. Early gestation as the critical time-window for changes in the prenatal environment to affect the adult human blood methylome. Int J Epidemiol. 2015, 44, 1211–1223. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Tian, F.; Tang, Z.; Song, G.; Pan, Y.; He, B.; Bao, Q. Loss of imprinting of IGF2 correlates with hypomethylation of the H19 differentially methylated region in the tumor tissue of colorectal cancer patients. Mol. Med. Rep. 2012, 5, 1536–1540. [Google Scholar] [CrossRef]
- Waterland, R.A.; Jirtle, R.L. Transposable Elements: Targets for Early Nutritional Effects on Epigenetic Gene Regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Vaiserman, A.; Lushchak, O. Prenatal famine exposure and adult health outcomes: An epigenetic link. Environ. Epigenetics 2021, 7, dvab013. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, M.B.; Firoz, A.S.; Imam, S.N.; Alzaman, N.; Samman, M.A. Epigenetics of human diseases and scope in future therapeutics. J. Taibah Univ. Med Sci. 2017, 12, 205–211. [Google Scholar] [CrossRef]
- Schaub, B.; Liu, J.; Höppler, S.; Schleich, I.; Huehn, J.; Olek, S.; Wieczorek, G.; Illi, S.; von Mutius, E. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J. Allergy Clin. Immunol. 2009, 123, 774–782.e5. [Google Scholar] [CrossRef]
- Wei, L.; Vahedi, G.; Sun, H.-W.; Watford, W.T.; Takatori, H.; Ramos, H.L.; Takahashi, H.; Liang, J.; Gutierrez-Cruz, G.; Zang, C.; et al. Discrete Roles of STAT4 and STAT6 Transcription Factors in Tuning Epigenetic Modifications and Transcription during T Helper Cell Differentiation. Immunity 2010, 32, 840–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, D.R.; Cho, E.; Copeland, T.D.; Guszczynski, T.; Yang, E.; Seth, A.K.; Farrar, W.L. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genom. Proteom. 2007, 4, 387–398. [Google Scholar]
- Zijlstra, G.J.; Hacken, N.H.T.T.; Hoffmann, R.F.; van Oosterhout, A.J.M.; Heijink, I.H. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur. Respir. J. 2012, 39, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Ogryzko, V.V.; Schiltz, R.; Russanova, V.; Howard, B.H.; Nakatani, Y. The Transcriptional Coactivators p300 and CBP Are Histone Acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Villagra, A.; Cheng, F.; Wang, H.-W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100, Erratum in Nat. Immunol. 2009, 10, 665. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Liu, J.; Wu, X.; Liu, S.; Li, G.; Han, C.; Song, L.; Li, Z.; Wang, Q.; Wang, J.; et al. Histone Methyltransferase Ash1l Suppresses Interleukin-6 Production and Inflammatory Autoimmune Diseases by Inducing the Ubiquitin-Editing Enzyme A20. Immunity 2013, 39, 470–481. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.B.; Rowell, E.; Sekimata, M. Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 2009, 9, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Ansel, K.M.; Lee, D.U.; Rao, A. An epigenetic view of helper T cell differentiation. Nat. Immunol. 2003, 4, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Kim, S.T.; Spilianakis, C.; Fields, P.E.; Flavell, R.A. T Helper Cell Differentiation: Regulation by cis Elements and Epigenetics. Immunity 2006, 24, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obata, Y.; Furusawa, Y.; Hase, K. Epigenetic modifications of the immune system in health and disease. Immunol. Cell Biol. 2015, 93, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-G.; Sahoo, A.; Im, S.-H. Epigenetic Regulation of Cytokine Gene Expression in T Lymphocytes. Yonsei Med J. 2009, 50, 322–330. [Google Scholar] [CrossRef] [Green Version]
- David, A.L.; McIntosh, J.; Peebles, D.M.; Cook, T.; Waddington, S.; Weisz, B.; Wigley, V.; Abi-Nader, K.; Boyd, M.; Davidoff, A.M.; et al. Recombinant Adeno-Associated Virus-Mediated In Utero Gene Transfer Gives Therapeutic Transgene Expression in the Sheep. Hum. Gene Ther. 2011, 22, 419–426. [Google Scholar] [CrossRef]
- Mattar, C.N.; Nathwani, A.C.; Waddington, S.N.; Dighe, N.; Kaeppel, C.; Nowrouzi, A.; Mcintosh, J.; Johana, N.B.; Ogden, B.; Fisk, N.M.; et al. Stable Human FIX Expression After 0.9G Intrauterine Gene Transfer of Self-complementary Adeno-associated Viral Vector 5 and 8 in Macaques. Mol. Ther. 2011, 19, 1950–1960. [Google Scholar] [CrossRef]
- Piedimonte, G.; Harford, T. Effects of maternal−fetal transmission of viruses and other environmental agents on lung development. Pediatr. Res. 2020, 87, 420–426. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Literature Authors (Year) (Ref#) | Species | Treatment of Pregnant Dam | Postnatal Treatment of Offspring | |||||
|---|---|---|---|---|---|---|---|---|
| First Stimulation (mg/kg BW) | Period (Gestational Day, GD) | Second Stimulation | Period (Postnatal Day, PD) | Findings | Histopathology | Assumed Pathogenesis | ||
| Charity et al. (2014) [68] | Mouse | Poly (I:C) (20) | Third trimester (GD12.5) | LPS and IFN-γ | PD 21 | Increase in IL-1, and IL12 in vitro | NE | Macrophage 1 polarization |
| Destanie et al. (2017) [67] | Monkey | Poly (I:C) (2500) | First and second trimester (GD 43,44, 46, 100,101, and 103) | LPS or Poly (I:C) | 1 year | Increase in IL-1, IL-2, IL4, IL6, IL12, and TNF-α in vitro | NE | Activation of innate and Th2 immune response |
| Shimizu et al. (2021) [66] | Mouse | Poly (I:C) (20) | Third trimester (GD 12.5, 14.5, and 16.5) | Poly (I:C) | PD 21-28 | Increase in IL-6, IL-17, and TNF-α in serum | Liver necrosis | Unfolded protein response defects |
| Literature Authors (Year) (Ref#) | Species | Treatment of Pregnant Dam | Postnatal Treatment of Offspring | |||||
|---|---|---|---|---|---|---|---|---|
| First Stimulation (μg/kg/dose) | Period (Gestational Day, GD) | Second Stimulation | Period (Postnatal Day, PD) | Findings | Histopathology | Assumed Pathogenesis | ||
| Lasaka et al. (2007) [83] | Rat | LPS (500) | Third trimester (GD 18) | LPS | PD 21 | Decrease in IL-1, IL-6, and TNF-α in serum | NE | ND |
| Surriga et al. (2009) [73] | Rat | LPS (500) | Third trimester (GD 18) | LPS | PD 21 | Decrease in IL6 mRNA expression in the liver | NE | Suppression of MAPK P42/44 |
| Basta-Kaim et al. (2012) [77] | Rat | LPS (1000) | Second to third trimester (Every 2days from GD 7) | Concanavalin A | PD 30 and 90 | Increase in IL-1β, IL-2, IL-6, and TNF-α in vitro | NE | Increased proliferative activity of splenocytes |
| Kirsten et al. (2013) [75] | Rat | LPS (100) | Second trimester (GD 9) | LPS | PD 60-67 | Increase in IL-1β in serum | NE | Glucocorticoid dysregulation |
| Zager et al. (2013) [85] | Mouse | LPS (120) | Third trimester (GD 17) | LPS | PD 70 | Increase in IL-12 in vitro | NE | Skewing of the cytokine balance towards Th1 |
| Hsueh et al. (2017) [75] | Mouse | LPS (25, 25, 50) | Third trimester (GD 15, 16 and 17) | LPS | PD 56 | Increase in IL-1, IL-6, IL-10, IL-12, IL-17, TNF-α, and IFN-γ in serum | NE | Increase in MCP-1 level |
| Adams et al. (2020) [82] | Mouse | LPS (10) | First to third trimester (GD 0, 7, 14) | LPS | PD 49 | Increase in IL-1, IL-6, and IL-10 mRNA expression in the spleen | NE | Glucocorticoid dysregulation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimizu, Y.; Sakata-Haga, H.; Saikawa, Y.; Hatta, T. Influence of Immune System Abnormalities Caused by Maternal Immune Activation in the Postnatal Period. Cells 2023, 12, 741. https://doi.org/10.3390/cells12050741
Shimizu Y, Sakata-Haga H, Saikawa Y, Hatta T. Influence of Immune System Abnormalities Caused by Maternal Immune Activation in the Postnatal Period. Cells. 2023; 12(5):741. https://doi.org/10.3390/cells12050741
Chicago/Turabian StyleShimizu, Yo, Hiromi Sakata-Haga, Yutaka Saikawa, and Toshihisa Hatta. 2023. "Influence of Immune System Abnormalities Caused by Maternal Immune Activation in the Postnatal Period" Cells 12, no. 5: 741. https://doi.org/10.3390/cells12050741
APA StyleShimizu, Y., Sakata-Haga, H., Saikawa, Y., & Hatta, T. (2023). Influence of Immune System Abnormalities Caused by Maternal Immune Activation in the Postnatal Period. Cells, 12(5), 741. https://doi.org/10.3390/cells12050741