

Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis

{kind=link}
{kind=link}
{kind=link}
Abstract
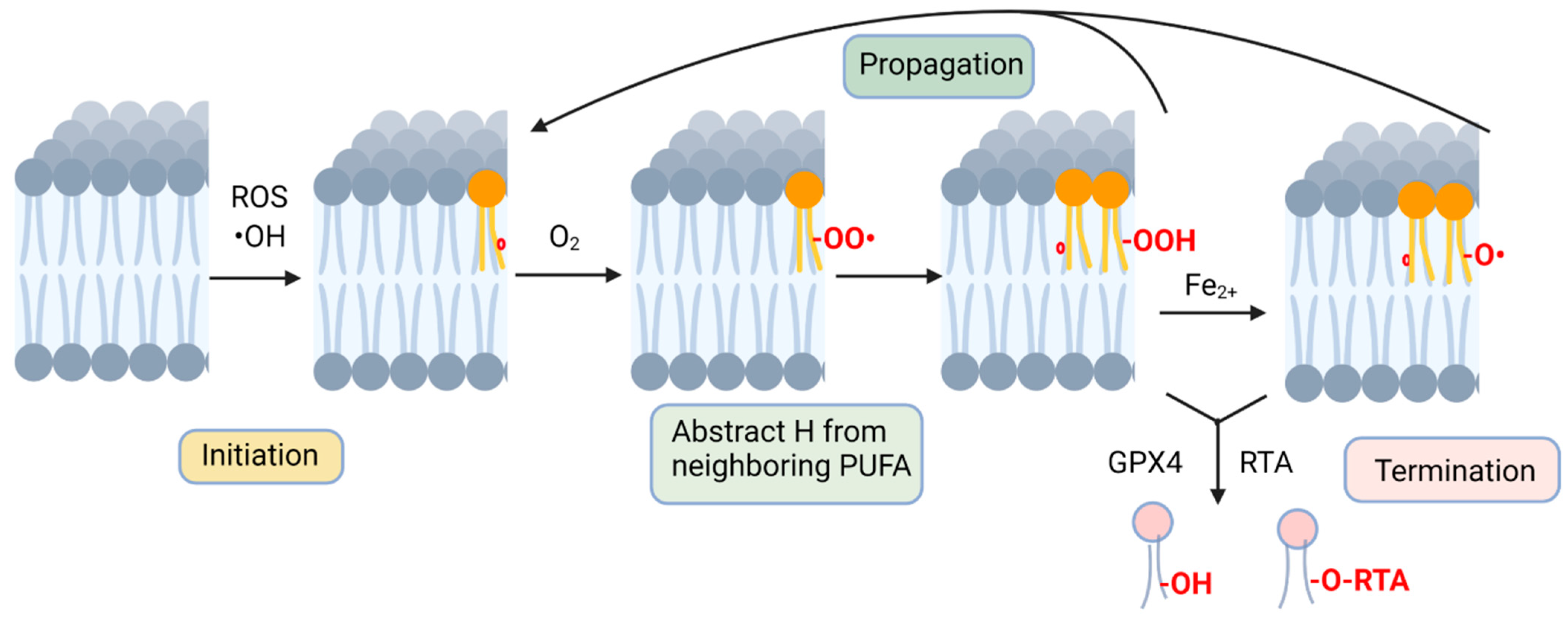
:1. Introduction
2. Lipid Peroxides Are Hallmarks of Ferroptosis
3. Non-Enzymatic Lipid Peroxidation
4. Radical-Trapping Antioxidants (RTA) Are Potent Ferroptosis Inhibitors
5. Iron Accumulation Leads to Ferroptosis in Aging Caenorhabditis elegans
6. Omega-6 PUFAs Promote Ferroptosis
7. Germ Cell Surveillance: A Physiological Role for Ferroptosis in C. elegans
8. Fatty Acid Composition of Ether Lipids Influences Ferroptosis
9. Monounsaturated Fatty Acids Protect Membranes from Ferroptosis
10. Lipid Remodeling Enzymes Influence Membrane Composition and Ferroptosis
11. The Ability of Specific PUFAs to Induce Ferroptosis May Depend on Enzymatic Conversion by Lipoxygenases or CYPs
12. DGLA-Induced Ferroptosis in C. elegans Mediated by CYP Activity
13. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, W.; Bae, K.-H.; Lee, S.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Jiang, X.; Wu, M.; Cao, X.; Bao, W.; Zhu, L.-Q. Ferroptosis, a Potential Therapeutic Target in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 704298. [Google Scholar] [CrossRef]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef]
- Mi, Y.; Gao, X.; Xu, H.; Cui, Y.; Zhang, Y.; Gou, X. The Emerging Roles of Ferroptosis in Huntington’s Disease. NeuroMolecular Med. 2019, 21, 110–119. [Google Scholar] [CrossRef]
- Li, H.; Lin, L.; Xia, Y.-L.; Xie, Y.; Yang, X. Research progress on the role of ferroptosis in cardiovascular disease. Front. Cardiovasc. Med. 2022, 9, 1077332. [Google Scholar] [CrossRef]
- Li, W.; Li, W.; Leng, Y.; Xiong, Y.; Xia, Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020, 39, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Jiang, C.; Mei, G.; Zhao, Y.; Chen, L.; Liu, J.; Tang, Y.; Gao, C.; Yao, P. Quercetin Alleviates Ferroptosis of Pancreatic β Cells in Type 2 Diabetes. Nutrients 2020, 12, 2954. [Google Scholar] [CrossRef]
- Hao, L.; Mi, J.; Song, L.; Guo, Y.; Li, Y.; Yin, Y.; Zhang, C. SLC40A1 Mediates Ferroptosis and Cognitive Dysfunction in Type 1 Diabetes. Neuroscience 2021, 463, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.-S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, S.; Kobayashi, Y.; Ide, K.; Strausser, S.A.; Abe, K.; Herbek, S.; O’Brien, L.L.; Crowley, S.D.; Barisoni, L.; Tata, A.; et al. Ferroptotic stress promotes the accumulation of pro-inflammatory proximal tubular cells in maladaptive renal repair. Elife 2021, 10, e68603. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, Y.; Jiang, R.; Xue, R.; Yin, X.; Wu, M.; Meng, Q. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, X.; Ren, Z.; Li, Y.; Zou, W.; Chen, J.; Wang, H. Overcoming cancer chemotherapy resistance by the induction of ferroptosis. Drug Resist. Updat. 2023, 66, 100916. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Mao, C.; Yan, Y.; Zhuang, L.; Gan, B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell 2021, 12, 836–857. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.P.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e421. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 5155. [Google Scholar] [CrossRef] [Green Version]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020, 22, 1042–1048. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress:4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [Green Version]
- Niedernhofer, L.J.; Daniels, J.S.; Rouzer, C.A.; Greene, R.E.; Marnett, L.J. Malondialdehyde, a Product of Lipid Peroxidation, Is Mutagenic in Human Cells. J. Biol. Chem. 2003, 278, 31426–31433. [Google Scholar] [CrossRef] [Green Version]
- Wagner, B.A.; Buettner, G.R.; Burns, C.P. Free Radical-Mediated Lipid Peroxidation in Cells: Oxidizability Is a Function of Cell Lipid bis-Allylic Hydrogen Content. Biochemistry 1994, 33, 4449–4453. [Google Scholar] [CrossRef] [PubMed]
- Vigor, C.; Bertrand-Michel, J.; Pinot, E.; Oger, C.; Vercauteren, J.; Le Faouder, P.; Galano, J.-M.; Lee, J.C.-Y.; Durand, T. Non-enzymatic lipid oxidation products in biological systems: Assessment of the metabolites from polyunsaturated fatty acids. J. Chromatogr. B 2014, 964, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A. An overview of lipid peroxidation with emphasis in outer segments of photoreceptors and the chemiluminescence assay. Int. J. Biochem. Cell Biol. 2006, 38, 1482–1495. [Google Scholar] [CrossRef]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [Green Version]
- Ingold, K.U.; Pratt, D.A. Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodencal, J.; Dixon, S.J. A tale of two lipids: Lipid unsaturation commands ferroptosis sensitivity. Proteomics 2022, 2100308. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Shah, R.; Margison, K.; Pratt, D.A. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem. Biol. 2017, 12, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Komm, B.S.; Kharode, Y.P.; Bodine, P.V.N.; Harris, H.A.; Miller, C.P.; Lyttle, C.R. Bazedoxifene Acetate: A Selective Estrogen Receptor Modulator with Improved Selectivity. Endocrinology 2005, 146, 3999–4008. [Google Scholar] [CrossRef] [Green Version]
- Conlon, M.; Poltorack, C.D.; Forcina, G.C.; Armenta, D.A.; Mallais, M.; Perez, M.A.; Wells, A.; Kahanu, A.; Magtanong, L.; Watts, J.L.; et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat. Chem. Biol. 2021, 17, 665–674. [Google Scholar] [CrossRef]
- Heird, W.C.; Lapillonne, A. The Role of Essential Fatty Acids in Development. Annu. Rev. Nutr. 2005, 25, 549–571. [Google Scholar] [CrossRef]
- Smit, E.N.; Muskiet, F.A.; Boersma, E.R. The possible role of essential fatty acids in the pathophysiology of malnutrition: A review. Prostaglandins Leukot. Essent. Fatty Acids 2004, 71, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. An Increase in the Omega-6/Omega-3 Fatty Acid Ratio Increases the Risk for Obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef] [Green Version]
- Van Blarigan, E.; Fuchs, C.S.; Niedzwiecki, D.; Ye, X.; Zhang, S.; Song, M.; Saltz, L.; Mayer, R.J.; Mowat, R.B.; Whittom, R.; et al. Long-chain omega-3 fatty acid and fish intake after colon cancer diagnosis and disease-free, recurrence-free, and overall survival in CALGB 89803 (Alliance). J. Clin. Oncol. 2017, 35, 585. [Google Scholar] [CrossRef]
- Perez, M.A.; Clostio, A.J.; Houston, I.R.; Ruiz, J.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Ether lipid deficiency disrupts lipid homeostasis leading to ferroptosis sensitivity. PLOS Genet. 2022, 18, e1010436. [Google Scholar] [CrossRef]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021, 33, 1701–1715. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.-J.; et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 32433–32442. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e366. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.L.; Browse, J. Dietary manipulation implicates lipid signaling in the regulation of germ cell mainte-nance in C. elegans. Dev. Biol. 2006, 292, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.M.; Deline, M.L.; Watts, J.L. Stress response pathways protect germ cells from omega-6 polyunsaturated fatty acid-mediated toxicity in Caenorhabditis elegans. Dev. Biol. 2013, 373, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditis elegans and Human Cancer Cells. Dev. Cell 2020, 54, 447–454.e444. [Google Scholar] [CrossRef]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Liu, D.; Gu, W.; Chu, B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021, 28, 2536–2551. [Google Scholar] [CrossRef]
- Shi, X.; Tarazona, P.; Brock, T.J.; Browse, J.; Feussner, I.; Watts, J.L. A Caenorhabditis elegans model for ether lipid biosynthesis and function. J. Lipid Res. 2016, 57, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e429. [Google Scholar] [CrossRef]
- Watts, J.L.; Ristow, M. Lipid and Carbohydrate Metabolism in Caenorhabditis elegans. Genetics 2017, 207, 413–446. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.L.; Browse, J. Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 5854–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Schroeder, E.A.; Silva-García, C.G.; Hebestreit, K.; Mair, W.B.; Brunet, A. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 2017, 544, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Lands, W.E.M. Stories about acyl chains. Biochim. Biophys. Acta 2000, 1483, 1–14. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, M.I.J.; Aichler, M.; Walch, M.A.A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Mueller, G.D.; Williams, K.J.; Billmann, M.; Chan, K.; Armenta, D.A.; Pope, L.E.; Moffat, J.; Boone, C.; Myers, C.L.; et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem. Biol. 2022, 29, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Central Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.-Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021, 38, 101744. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [Green Version]
- Konkel, A.; Schunck, W.-H. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim. Biophys. Acta 2011, 1814, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Sarparast, M.; Dattmore, D.; Alan, J.; Lee, K.S.S. Cytochrome P450 Metabolism of Polyunsaturated Fatty Acids and Neurodegeneration. Nutrients 2020, 12, 3523. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2017, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef]
- Kosel, M.; Wild, W.; Bell, A.; Rothe, M.; Lindschau, C.; Steinberg, C.E.W.; Schunck, W.-H.; Menzel, R. Eicosanoid formation by a cytochrome P450 isoform expressed in the pharynx of Caenorhabditis elegans. Biochem. J. 2011, 435, 689–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deline, M.; Keller, J.; Rothe, M.; Schunck, W.-H.; Menzel, R.; Watts, J.L. Epoxides Derived from Dietary Dihomo-Gamma-Linolenic Acid Induce Germ Cell Death in C. elegans. Sci. Rep. 2015, 5, 15417. [Google Scholar] [CrossRef] [Green Version]
- Sarparast, M.; Pourmand, E.; Hinman, J.; Vonarx, D.; Reason, T.; Zhang, F.; Paithankar, S.; Chen, B.; Borhan, B.; Watts, J.L.; et al. Dihydroxy-Metabolites of Dihomo-gamma-linolenic Acid Drive Ferroptosis-Mediated Neurodegeneration. bioRxiv 2023. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. https://doi.org/10.3390/cells12050804
Mortensen MS, Ruiz J, Watts JL. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells. 2023; 12(5):804. https://doi.org/10.3390/cells12050804
Chicago/Turabian StyleMortensen, Michael S., Jimena Ruiz, and Jennifer L. Watts. 2023. "Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis" Cells 12, no. 5: 804. https://doi.org/10.3390/cells12050804
APA StyleMortensen, M. S., Ruiz, J., & Watts, J. L. (2023). Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells, 12(5), 804. https://doi.org/10.3390/cells12050804