

Differences in Cerebral Glucose Metabolism in ALS Patients with and without C9orf72 and SOD1 Mutations

 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.1.1. Healthy Controls
2.1.2. Patients
2.2. Propensity Score Matching
2.3. Assessment of Clinical Functioning
2.4. Image Analysis
2.4.1. PET Acquisition and Reconstruction
2.4.2. FDG PET Image Analysis
2.5. Statistical Analysis
3. Results
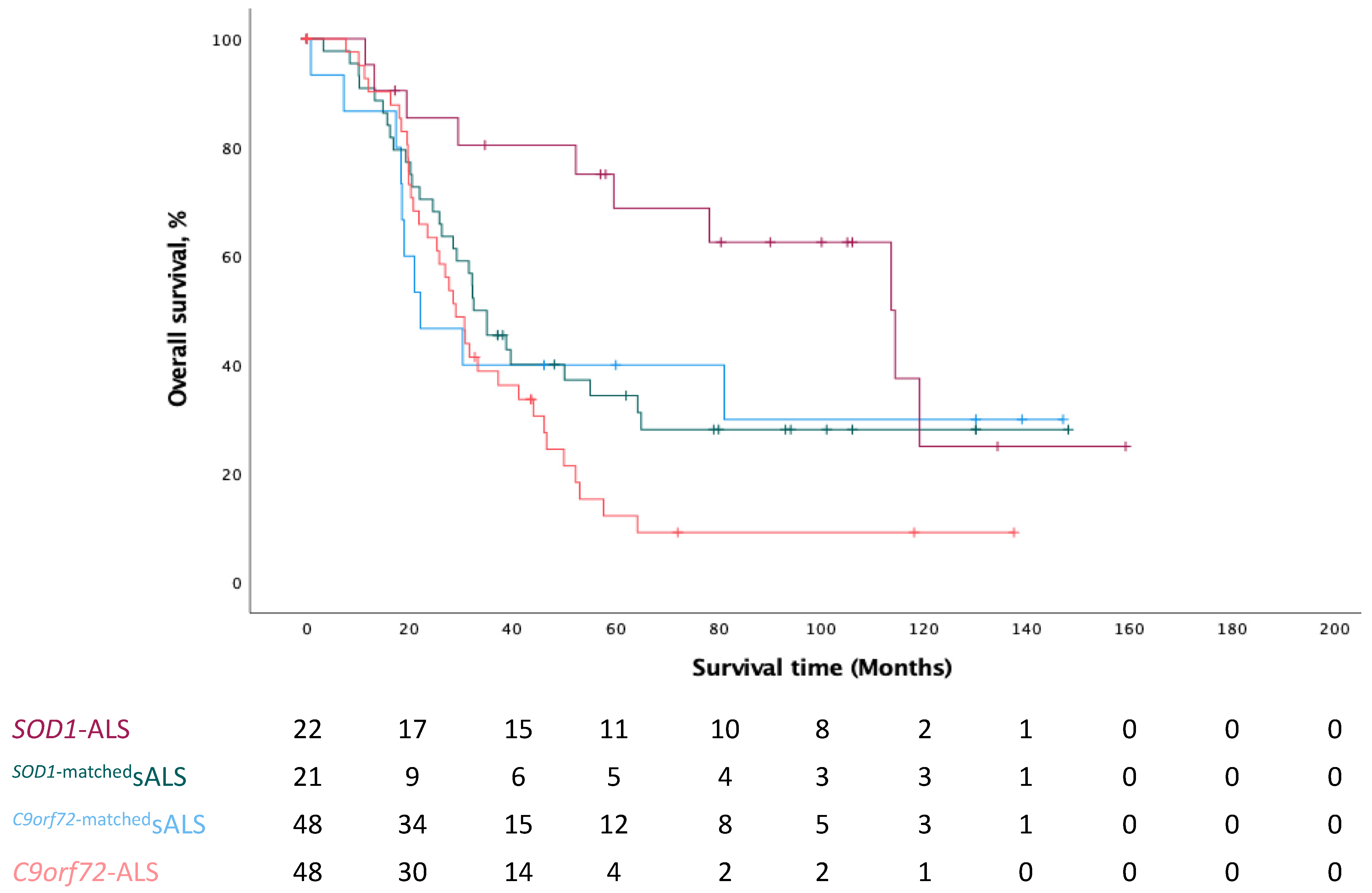
3.1. Demographics and Clinical Characteristics
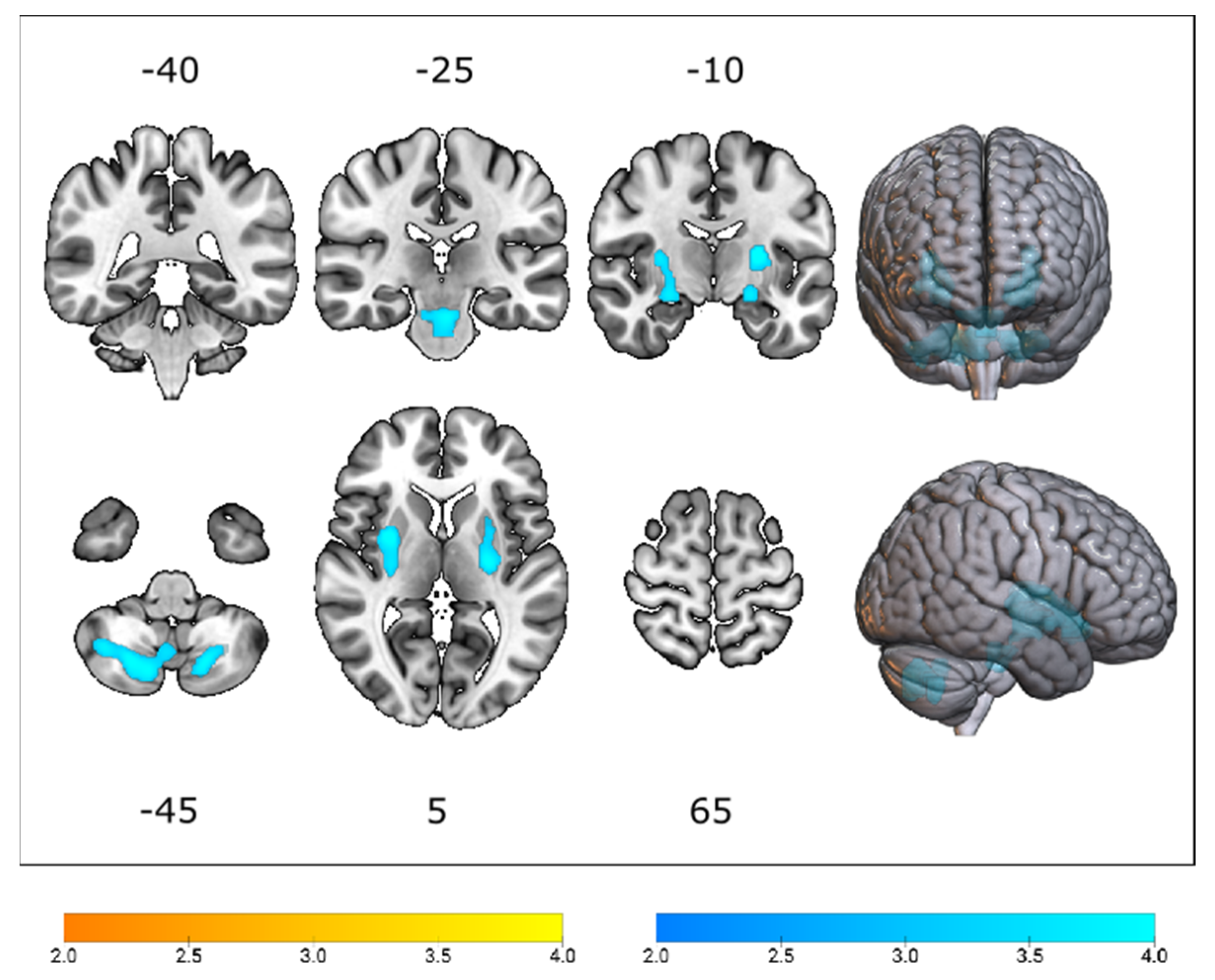
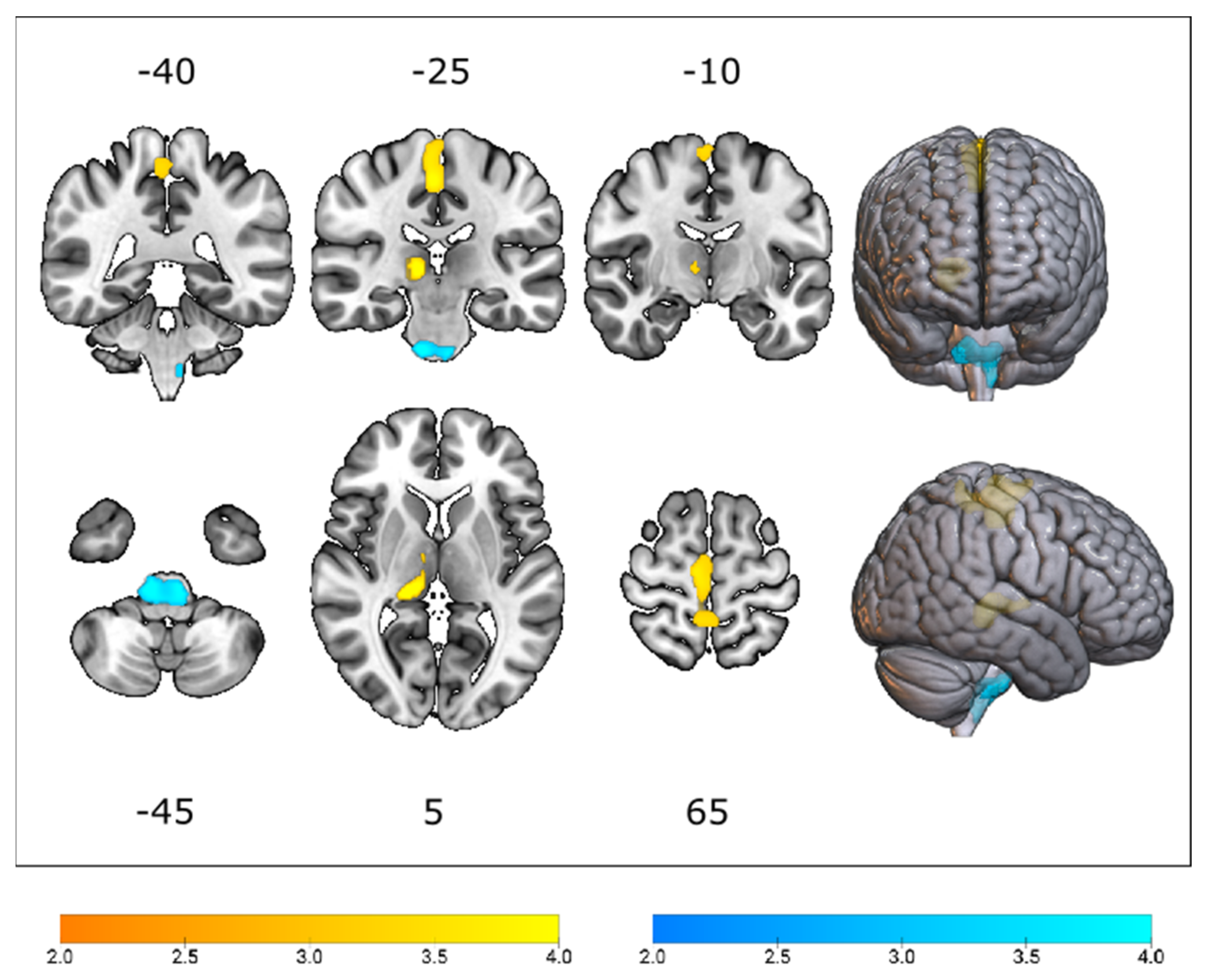
3.2. Cerebral Glucose Metabolic Changes between Different Genotypes
3.3. Differences in Cerebral Glucose Metabolism between ALS Groups and Healthy Controls
3.4. Effect of Genetic Status on Cerebral 18F FDG Uptake
4. Discussion
4.1. The Effect of C9orf72 Status on Cerebral Glucose Metabolism
4.2. The Effect of SOD1 Status on Cerebral Glucose Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper-Knock, J.; Kirby, J.; Highley, R.; Shaw, P.J. The Spectrum of C9orf72-mediated Neurodegeneration and Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 326–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper-Knock, J.; Shaw, P.J.; Kirby, J. The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol. 2014, 127, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Snowden, J.S.; Rollinson, S.; Thompson, J.C.; Harris, J.M.; Stopford, C.L.; Richardson, A.M.; Jones, M.; Gerhard, A.; Davidson, Y.S.; Robinson, A.; et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012, 135 Pt 3, 693–708. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M.; Forsgren, L.; Binzer, M.; Nilsson, P.; Ala-Hurula, V.; Keränen, M.L.; Bergmark, L.; Saarinen, A.; Haltia, T.; Tarvainen, I.; et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain 1996, 119 Pt 4, 1153–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.F.; Wu, Z.Y. Genotype-phenotype correlations of amyotrophic lateral sclerosis. Transl. Neurodegener. 2016, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Juneja, T.; Pericak-Vance, M.A.; Laing, N.G.; Dave, S.; Siddique, T. Prognosis in familial amyotrophic lateral sclerosis: Progression and survival in patients with glu100gly and ala4val mutations in Cu,Zn superoxide dismutase. Neurology 1997, 48, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Salachas, F.; Cazeneuve, C.; Gordon, P.; Bricka, B.; Camuzat, A.; Guillot-Noël, L.; Russaouen, O.; Bruneteau, G.; Pradat, P.F.; et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Project MinE: Study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 2018, 26, 1537–1546. [CrossRef] [Green Version]
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Bella, E.D.; Bersano, E.; Bruzzone, M.G.; Gellera, C.; Pensato, V.; Lauria, G.; Consonni, M. Behavioral and Cognitive Phenotypes of Patients With Amyotrophic Lateral Sclerosis Carrying SOD1 Variants. Neurology 2022, 99, e2052–e2062. [Google Scholar] [CrossRef]
- Trojsi, F.; Siciliano, M.; Femiano, C.; Santangelo, G.; Lunetta, C.; Calvo, A.; Moglia, C.; Marinou, K.; Ticozzi, N.; Drago Ferrante, G.; et al. Comorbidity of dementia with amyotrophic lateral sclerosis (ALS): Insights from a large multicenter Italian cohort. J. Neurol. 2017, 264, 2224–2231. [Google Scholar] [CrossRef]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Van Laere, K.; Vanhee, A.; Verschueren, J.; De Coster, L.; Driesen, A.; Dupont, P.; Robberecht, W.; Van Damme, P. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: A prospective study. JAMA Neurol. 2014, 71, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Van Weehaeghe, D.; Devrome, M.; Schramm, G.; De Vocht, J.; Deckers, W.; Baete, K.; Van Damme, P.; Koole, M.; Van Laere, K. Combined brain and spinal FDG PET allows differentiation between ALS and ALS mimics. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2681–2690. [Google Scholar] [CrossRef]
- Van Weehaeghe, D.; Ceccarini, J.; Delva, A.; Robberecht, W.; Van Damme, P.; Van Laere, K. Prospective Validation of 18F-FDG Brain PET Discriminant Analysis Methods in the Diagnosis of Amyotrophic Lateral Sclerosis. J. Nucl. Med. 2016, 57, 1238–1243. [Google Scholar] [CrossRef] [Green Version]
- Pagani, M.; Chiò, A.; Valentini, M.C.; Öberg, J.; Nobili, F.; Calvo, A.; Moglia, C.; Bertuzzo, D.; Morbelli, S.; De Carli, F.; et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology 2014, 83, 1067–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canosa, A.; Pagani, M.; Cistaro, A.; Montuschi, A.; Iazzolino, B.; Fania, P.; Cammarosano, S.; Ilardi, A.; Moglia, C.; Calvo, A.; et al. 18F-FDG-PET correlates of cognitive impairment in ALS. Neurology 2016, 86, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cistaro, A.; Pagani, M.; Montuschi, A.; Calvo, A.; Moglia, C.; Canosa, A.; Restagno, G.; Brunetti, M.; Traynor, B.J.; Nobili, F.; et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 844–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönecker, S.; Neuhofer, C.; Otto, M.; Ludolph, A.; Kassubek, J.; Landwehrmeyer, B.; Anderl-Straub, S.; Semler, E.; Diehl-Schmid, J.; Prix, C.; et al. Atrophy in the Thalamus But Not Cerebellum Is Specific for C9orf72 FTD and ALS Patients—An Atlas-Based Volumetric MRI Study. Front. Aging Neurosci. 2018, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Diehl-Schmid, J.; Licata, A.; Goldhardt, O.; Förstl, H.; Yakushew, I.; Otto, M.; Anderl-Straub, S.; Beer, A.; Ludolph, A.C.; Landwehrmeyer, G.B.; et al. FDG-PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9ORF72 mutations. Transl. Psychiatry 2019, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Canosa, A.; Calvo, A.; Moglia, C.; Vasta, R.; Palumbo, F.; Solero, L.; Di Pede, F.; Cabras, S.; Arena, V.; Zocco, G.; et al. Amyotrophic lateral sclerosis with SOD1 mutations shows distinct brain metabolic changes. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 2242–2250. [Google Scholar] [CrossRef]
- Schrooten, M.; Smetcoren, C.; Robberecht, W.; Van Damme, P. Benefit of the Awaji diagnostic algorithm for amyotrophic lateral sclerosis: A prospective study. Ann. Neurol. 2011, 70, 79–83. [Google Scholar] [CrossRef]
- Rosenbaum, P.R.; Rubin, D.B. The central role of the propensity score in observational studies for causal effects. Biometrika 1983, 70, 41–55. [Google Scholar] [CrossRef]
- D’Agostino, R.B., Jr. Propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Stat. Med. 1998, 17, 2265–2281. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Tzourio-Mazoyer, N.; Landeau, B.; Papathanassiou, D.; Crivello, F.; Etard, O.; Delcroix, N.; Mazoyer, B.; Joliot, M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 2002, 15, 273–289. [Google Scholar] [CrossRef]
- Rolls, E.T.; Huang, C.C.; Lin, C.P.; Feng, J.; Joliot, M. Automated anatomical labelling atlas 3. Neuroimage 2020, 206, 116189. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- De Vocht, J.; Blommaert, J.; Devrome, M.; Radwan, A.; Van Weehaeghe, D.; De Schaepdryver, M.; Ceccarini, J.; Rezaei, A.; Schramm, G.; van Aalst, J.; et al. Use of Multimodal Imaging and Clinical Biomarkers in Presymptomatic Carriers of C9orf72 Repeat Expansion. JAMA Neurol. 2020, 77, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Starr, A.; Sattler, R. Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 2018, 1693 Pt A, 98–108. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Tzioras, M.; Paolicelli, R.C. Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration. Front. Cell. Neurosci. 2019, 13, 63. [Google Scholar] [CrossRef] [Green Version]
- Calió, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial Dysfunction, Neurogenesis, and Epigenetics: Putative Implications for Amyotrophic Lateral Sclerosis Neurodegeneration and Treatment. Front. Neurosci. 2020, 14, 679. [Google Scholar] [CrossRef]
- Terada, T.; Therriault, J.; Kang, M.S.; Savard, M.; Pascoal, T.A.; Lussier, F.; Tissot, C.; Wang, Y.-T.; Benedet, A.; Poltronetti, N.M.; et al. Mitochondrial complex I abnormalities underlie neurodegeneration and cognitive decline in Alzheimer’s disease. Eur. J. Neurol. 2022, 29, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Malpetti, M.; Holland, N.; Jones, P.S.; Ye, R.; Cope, T.E.; Fryer, T.D.; Hong, Y.T.; Savulich, G.; Rittman, T.; Passamonti, L.; et al. Synaptic density in carriers of C9orf72 mutations: A [11C]UCB-J PET study. Ann. Clin. Transl. Neurol. 2021, 8, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Cistaro, A.; Valentini, M.C.; Chiò, A.; Nobili, F.; Calvo, A.; Moglia, C.; Montuschi, A.; Morbelli, S.; Salmaso, D.; Fania, P.; et al. Brain hypermetabolism in amyotrophic lateral sclerosis: A FDG PET study in ALS of spinal and bulbar onset. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Matías-Guiu, J.A.; Pytel, V.; Cabrera-Martín, M.N.; Galán, L.; Valles-Salgado, M.; Guerrero, A.; Moreno-Ramos, T.; Matías-Guiu, J.; Carreras, J.L. Amyloid- and FDG-PET imaging in amyotrophic lateral sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2050–2060. [Google Scholar] [CrossRef]
- Brettschneider, J.; Toledo, J.B.; Van Deerlin, V.M.; Elman, L.; McCluskey, L.; Lee, V.M.; Trojanowski, J.Q. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39216. [Google Scholar] [CrossRef] [Green Version]
- Chew, S.; Atassi, N. Positron Emission Tomography Molecular Imaging Biomarkers for Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Daughrity, L.M.; Heckman, M.G.; Diehl, N.N.; Wuu, J.; Miller, T.M.; Pastor, P.; Trojanowski, J.Q.; Grossman, M.; Berry, J.D.; et al. Phosphorylated neurofilament heavy chain: A biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann. Neurol. 2017, 82, 139–146. [Google Scholar] [CrossRef]
- Irwin, D.J.; McMillan, C.T.; Brettschneider, J.; Libon, D.J.; Powers, J.; Rascovsky, K.; Toledo, J.B.; Boller, A.; Bekisz, J.; Chandrasekaran, K. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umoh, M.E.; Fournier, C.; Li, Y.; Polak, M.; Shaw, L.; Landers, J.E.; Hu, W.; Gearing, M.; Glass, J.D. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016, 87, 1024–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanovello, M.; Sorarù, G.; Campi, C.; Anglani, M.; Spimpolo, A.; Berti, S.; Bussè, C.; Mozzetta, S.; Cagnin, A.; Cecchin, D. Brain Stem Glucose Hypermetabolism in Amyotrophic Lateral Sclerosis/Frontotemporal Dementia and Shortened Survival: An 18F-FDG PET/MRI Study. J. Nucl. Med. 2022, 63, 777–784. [Google Scholar] [CrossRef]
- Devrome, M.; Van Weehaeghe, D.; De Vocht, J.; Van Damme, P.; Van Laere, K.; Koole, M. Glucose metabolic brain patterns to discriminate amyotrophic lateral sclerosis from Parkinson plus syndromes. EJNMMI Res. 2018, 8, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Weehaeghe, D.; Ceccarini, J.; Willekens, S.M.; de Vocht, J.; van Damme, P.; van Laere, K. Is there a glucose metabolic signature of spreading TDP-43 pathology in amyotrophic lateral sclerosis? Q. J. Nucl. Med. Mol. Imaging 2020, 64, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Sideris, D.I.; Carroll, E.; Rotariu, S.; Salomonsson, S.; Tzioras, M.; McKenzie, C.A.; Smith, C.; von Arnim, C.A.F.; Ludolph, A.C.; et al. Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Mochizuki, Y.; Takeuchi, R.; Shimizu, T.; Nagao, M.; Watabe, K.; Arai, N.; Oyanagi, K.; Onodera, O.; Hayashi, M.; et al. Clinicopathological characteristics of patients with amyotrophic lateral sclerosis resulting in a totally locked-in state (communication Stage V). Acta Neuropathol. Commun. 2016, 4, 107. [Google Scholar] [CrossRef] [Green Version]
- Agosta, F.; Spinelli, E.G.; Marjanovic, I.V.; Stevic, Z.; Pagani, E.; Valsasina, P.; Salak-Djokic, B.; Jankovic, M.; Lavrnic, D.; Kostic, V.S.; et al. Unraveling ALS due to SOD1 mutation through the combination of brain and cervical cord MRI. Neurology 2018, 90, e707. [Google Scholar] [CrossRef]
- Turner, M.R.; Hammers, A.; Allsop, J.; Al-chalabi, A.; Shaw, C.E.; Brooks, D.J.; Nigel Leigh, P.; Andersen, P.M. Volumetric cortical loss in sporadic and familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2007, 8, 343–347. [Google Scholar] [CrossRef]
- Turner, M.R.; Hammers, A.; Al-Chalabi, A.; Shaw, C.E.; Andersen, P.M.; Brooks, D.J.; Leigh, P.N. Distinct cerebral lesions in sporadic and ‘D90A’ SOD1 ALS: Studies with [11C]flumazenil PET. Brain 2005, 128, 1323–1329. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.R.; Rabiner, E.A.; Al-Chalabi, A.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Andersen, P.M. Cortical 5-HT1A receptor binding in patients with homozygous D90A SOD1 vs sporadic ALS. Neurology 2007, 68, 1233–1235. [Google Scholar] [CrossRef]
- Tondo, G.; Iaccarino, L.; Cerami, C.; Vanoli, G.E.; Presotto, L.; Masiello, V.; Coliva, A.; Salvi, F.; Bartolomei, I.; Mosca, L.; et al. (11) C-PK11195 PET-based molecular study of microglia activation in SOD1 amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1513–1523. [Google Scholar] [CrossRef]
- Van Weehaeghe, D.; Babu, S.; De Vocht, J.; Zürcher, N.R.; Chew, S.; Tseng, C.J.; Loggia, M.L.; Koole, M.; Rezaei, A.; Schramm, G.; et al. Moving Toward Multicenter Therapeutic Trials in Amyotrophic Lateral Sclerosis: Feasibility of Data Pooling Using Different Translocator Protein PET Radioligands. J. Nucl. Med. 2020, 61, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.E.; Imai, K.; King, G.; Stuart, E.A. Matching as Nonparametric Preprocessing for Reducing Model Dependence in Parametric Causal Inference. Political Anal. 2007, 15, 199–236. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Md (IQR) | C9orf72-ALS | C9orf72-matchedsALS | SOD1-ALS | SOD1-matchedsALS | Test Statistic | p-Value |
|---|---|---|---|---|---|---|
| Age at FDG PET (Yr) | 58.5 (9.00) | 63 (16.75) | 54.5 (20.00) | 60 (22.50) | χ2(4): 4.159 | 0.385 |
| Diagnostic Delay (Yr) | 0.73 (0.62) | 0.34 (0.22) | 1.51 (1.48) | 0.43 (0.27) | χ2(3): 47.322 | <0.001 |
| Md (95% CI) | ||||||
| Survival Time (M) | 28.93 (24.00–33.86) | 32.43 (24.75–40.11) | 114.43 (71.02–157.84) | 22.07 (7.72–36.42) | χ2(3): 11.130 | 0.011 |
| n (%) | ||||||
| Sex: | ||||||
| 28 (58) | 32 (67) | 12 (55) | 13 (62) | χ2: 1.184 | 0.757 |
| King Stage: | ||||||
| 14 (29) | 18 (38) | 7 (32) | 8 (38) | χ2(3): 1.025 | 0.795 |
| 17 (35) | 12 (25) | 7 (32) | 8 (38) | ||
| 16 (33) | 16 (33) | 7 (32) | 5 (24) | ||
| 1 (2) | 2 (4) | 1 (5) | 0 (0) | ||
| Onset type: | ||||||
| 33 (69) | 31 (65) | 18 (82) | 14 (67) | χ2: 2.185 | 0.535 |
| Median (IQR) | SOD1-ALS | C9orf72-ALS | Test Statistic | pFDR Value |
|---|---|---|---|---|
| Mid Frontal gyrus | 1.100 (0.07) | 1.070 (0.07) | 403.5 | 0.248 |
| Precentral gyrus | 1.015 (0.04) | 0.990 (0.05) | 272.0 | 0.007 |
| Straight gyrus | 0.975 (0.05) | 0.970 (0.06) | 479.5 | 0.711 |
| Orbitofrontal cortex | 1.000 (0.07) | 1.010 (0.06) | 485.0 | 0.746 |
| Inferior frontal gyrus | 1.075 (0.06) | 1.050 (0.06) | 373.0 | 0.181 |
| Superior frontal gyrus | 1.010 (0.05) | 0.990 (0.04) | 384.0 | 0.165 |
| Medial orbital gyrus | 0.960 (0.05) | 0.970 (0.07) | 442.5 | 0.490 |
| Lateral orbital gyrus | 1.025 (0.05) | 1.020 (0.06) | 479.0 | 0.732 |
| Posterior orbital gyrus | 0.940 (0.03) | 0.960 (0.06) | 452.0 | 0.515 |
| Subgenual frontal cortex | 0.830 (0.05) | 0.835 (0.08) | 509.0 | 0.855 |
| Subcallosal area | 0.825 (0.05) | 0.800 (0.10) | 427.0 | 0.389 |
| Pre-subgenual frontal cortex | 1.030 (0.06) | 1.005 (0.12) | 515.0 | 0.893 |
| Hippocampus | 0.770 (0.18) | 0.780 (0.08) | 486.5 | 0.739 |
| Amygdala | 0.720 (0.07) | 0.745 (0.07) | 379.5 | 0.168 |
| Anterior temporal lobe (med) | 0.740 (0.04) | 0.755 (0.04) | 398.5 | 0.229 |
| Lateral Anterior temporal lobe | 0.860 (0.06) | 0.870 (0.04) | 505.5 | 0.896 |
| Parahippocampal and ambient gyri | 0.780 (0.03) | 0.790 (0.03) | 416.0 | 0.317 |
| Superior temporal gyrus, posterior | 1.010 (0.08) | 1.000 (0.06) | 469.5 | 0.652 |
| Middle and inferior temporal gyrus | 0.940 (0.04) | 0.940 (0.04) | 506.0 | 0.875 |
| Fusiform gyrus | 0.840 (0.04) | 0.840 (0.06) | 506.0 | 0.875 |
| Posterior temporal lobe | 1.000 (0.03) | 1.010 (0.03) | 457.5 | 0.543 |
| Superior temporal gyrus, anterior | 0.840 (0.05) | 0.820 (0.05) | 449.5 | 0.537 |
| Postcentral gyrus | 1.010 (0.03) | 0.980 (0.04) | 246.5 | 0.006 |
| Superior parietal gyrus | 1.090 (0.05) | 1.070 (0.04) | 374.5 | 0.172 |
| Inferolateral remainder parietal lobe | 1.055 (0.04) | 1.040 (0.04) | 383.5 | 0.174 |
| Lateral remainder of the occipital lobe | 1.100 (0.05) | 1.120 (0.07) | 438.0 | 0.468 |
| Lingual gyrus | 1.180 (0.06) | 1.190 (0.12) | 450.5 | 0.524 |
| Cuneus | 1.220 (0.07) | 1.255 (0.12) | 487.0 | 0.720 |
| Thalamus | 0.935 (0.06) | 0.880 (0.10) | 278.5 | 0.012 |
| Insula | 0.930 (0.04) | 0.925 (0.03) | 515.5 | 0.874 |
| Cingulate gyrus, anterior | 1.005 (0.06) | 0.975 (0.08) | 376.0 | 0.167 |
| Cingulate gyrus, posterior | 1.160 (0.05) | 1.120 (0.05) | 304.0 | 0.019 |
| Cerebellum | 0.960 (0.09) | 1.025 (0.07) | 264.0 | 0.008 |
| Medulla | 0.715 (0.10) | 0.790 (0.07) | 250.0 | 0.005 |
| Midbrain | 0.740 (0.09) | 0.810 (0.08) | 288.0 | 0.012 |
| Pons | 0.625 (0.06) | 0.680 (0.08) | 303.0 | 0.019 |
| Lentiform nucleus | 1.080 (0.07) | 1.155 (0.08) | 214.0 | 0.003 |
| Median (IQR) | C9orf72-ALS | C9orf72-matchedsALS | Test Statistic | pFDR-Value |
|---|---|---|---|---|
| Mid Frontal gyrus | 1.070 (0.07) | 1.085 (0.06) | 1070.0 | 0.675 |
| Precentral gyrus | 0.990 (0.05) | 1.010 (0.04) | 797.0 | 0.048 |
| Straight gyrus | 0.970 (0.06) | 0.950 (0.08) | 1377.0 | 0.242 |
| Orbitofrontal cortex | 1.010 (0.06) | 1.000 (0.07) | 1419.0 | 0.142 |
| Inferior frontal gyrus | 1.050 (0.06) | 1.055 (0.06) | 1041.5 | 0.550 |
| Superior frontal gyrus | 0.990 (0.04) | 1.000 (0.06) | 997.5 | 0.412 |
| Medial orbital gyrus | 0.970 (0.07) | 0.960 (0.04) | 1429.5 | 0.138 |
| Lateral orbital gyrus | 1.020 (0.06) | 1.000 (0.07) | 1321.0 | 0.377 |
| Posterior orbital gyrus | 0.960 (0.06) | 0.945 (0.06) | 1300.5 | 0.407 |
| Subgenual frontal cortex | 0.835 (0.08) | 0.820 (0.08) | 1343.0 | 0.314 |
| Subcallosal area | 0.800 (0.10) | 0.810 (0.10) | 1133.0 | 0.940 |
| Pre-subgenual frontal cortex | 1.005 (0.12) | 1.015 (0.12) | 1152.0 | 1.000 |
| Hippocampus | 0.780 (0.08) | 0.770 (0.09) | 1231.5 | 0.668 |
| Amygdala | 0.745 (0.07) | 0.720 (0.07) | 1368.0 | 0.261 |
| Ant. Temporal lobe (med) | 0.755 (0.04) | 0.730 (0.06) | 1497.0 | 0.263 |
| Ant. Temporal lobe (lateral) | 0.870 (0.04) | 0.860 (0.05) | 1320.5 | 0.362 |
| Parahippocampal and ambient gyri | 0.790 (0.03) | 0.790 (0.05) | 1272.5 | 0.513 |
| Superior temporal gyrus, posterior | 1.000 (0.06) | 1.015 (0.06) | 1008.5 | 0.414 |
| Middle and inferior temporal gyrus | 0.940 (0.04) | 0.930 (0.03) | 1362.0 | 0.182 |
| Fusiform gyrus | 0.840 (0.06) | 0.830 (0.05) | 1261.0 | 0.538 |
| Posterior temporal lobe | 1.010 (0.03) | 1.010 (0.04) | 1206.0 | 0.751 |
| Superior temporal gyrus, anterior | 0.820 (0.05) | 0.810 (0.08) | 1170.0 | 0.920 |
| Postcentral gyrus | 0.980 (0.04) | 1.010 (0.05) | 670.0 | 0.007 |
| Superior parietal gyrus | 1.070 (0.04) | 1.085 (0.04) | 774.0 | 0.031 |
| Inferolateral remainder parietal lobe | 1.040 (0.04) | 1.050 (0.03) | 922.5 | 0.241 |
| Lateral remainder occipital lobe | 1.120 (0.07) | 1.100 (0.06) | 1340.0 | 0.309 |
| Lingual gyrus | 1.190 (0.12) | 1.190 (0.11) | 1217.0 | 0.710 |
| Cuneus | 1.255 (0.12) | 1.230 (0.09) | 1227.0 | 0.673 |
| Thalamus | 0.880 (0.10) | 0.930 (0.09) | 724.5 | 0.019 |
| Insula | 0.925 (0.03) | 0.920 (0.06) | 1306.0 | 0.395 |
| Cingulate gyrus, ant. | 0.975 (0.08) | 1.005 (0.09) | 957.0 | 0.312 |
| Cingulate gyrus, post. | 1.120 (0.05) | 1.150 (0.05) | 659.0 | 0.011 |
| Cerebellum | 1.025 (0.07) | 0.985 (0.07) | 1482.5 | 0.062 |
| Medulla | 0.790 (0.07) | 0.740 (0.07) | 1602.5 | 0.012 |
| Midbrain | 0.810 (0.08) | 0.790 (0.07) | 1453.0 | 0.100 |
| Pons | 0.680 (0.08) | 0.650 (0.07) | 1421.5 | 0.148 |
| Lentiform nucleus | 1.155 (0.08) | 1.110 (0.09) | 1569.5 | 0.015 |
| Median (IQR) | SOD1-ALS | SOD1-matched sALS | Test Statistic | pFDR Value |
|---|---|---|---|---|
| Mid Frontal gyrus | 1.100 (0.07) | 1.090 (0.03) | 249.5 | 1.000 |
| Precentral gyrus | 1.015 (0.04) | 1.000 (0.04) | 299.0 | 1.000 |
| Straight gyrus | 0.975 (0.05) | 0.990 (0.06) | 207.0 | 1.000 |
| Orbitofrontal cortex | 1.000 (0.07) | 1.000 (0.06) | 214.0 | 0.965 |
| Inferior frontal gyrus | 1.075 (0.06) | 1.050 (0.06) | 265.0 | 1.000 |
| Superior frontal gyrus | 1.010 (0.05) | 1.010 (0.05) | 221.0 | 0.933 |
| Medial orbital gyrus | 0.960 (0.05) | 0.960 (0.05) | 208.0 | 1.000 |
| Lateral orbital gyrus | 1.025 (0.05) | 1.010 (0.06) | 260.5 | 1.000 |
| Posterior orbital gyrus | 0.940 (0.03) | 0.960 (0.05) | 195.5 | 1.000 |
| Subgenual frontal cortex | 0.830 (0.05) | 0.830 (0.05) | 243.5 | 0.937 |
| Subcallosal area | 0.825 (0.05) | 0.820 (0.07) | 229.0 | 0.988 |
| Pre-subgenual frontal cortex | 1.030 (0.06) | 1.040 (0.07) | 176.0 | 1.000 |
| Hippocampus | 0.770 (0.18) | 0.770 (0.06) | 221.0 | 0.905 |
| Amygdala | 0.720 (0.07) | 0.730 (0.05) | 207.5 | 1.000 |
| Anterior temporal lobe (med) | 0.740 (0.04) | 0.750 (0.05) | 200.0 | 1.000 |
| Lateral Anterior temporal lobe | 0.860 (0.06) | 0.870 (0.05) | 222.5 | 0.910 |
| Parahippocampal and ambient gyri | 0.780 (0.03) | 0.780 (0.05) | 227.0 | 0.975 |
| Superior temporal gyrus, posterior | 1.010 (0.08) | 1.010 (0.04) | 219.5 | 0.930 |
| Middle and inferior temporal gyrus | 0.940 (0.04) | 0.950 (0.06) | 212.0 | 1.000 |
| Fusiform gyrus | 0.840 (0.04) | 0.830 (0.05) | 252.5 | 1.000 |
| Posterior temporal lobe | 1.000 (0.03) | 1.020 (0.03) | 210.5 | 1.000 |
| Superior temporal gyrus, anterior | 0.840 (0.05) | 0.830 (0.06) | 193.5 | 1.000 |
| Postcentral gyrus | 1.010 (0.03) | 1.010 (0.04) | 276.5 | 1.000 |
| Superior parietal gyrus | 1.090 (0.05) | 1.070 (0.06) | 286.0 | 1.000 |
| Inferolateral remainder parietal lobe | 1.055 (0.04) | 1.050 (0.03) | 278.0 | 1.000 |
| Lateral remainder of the occipital lobe | 1.100 (0.05) | 1.100 (0.07) | 247.5 | 0.908 |
| Lingual gyrus | 1.180 (0.06) | 1.190 (0.07) | 230.0 | 0.981 |
| Cuneus | 1.220 (0.07) | 1.210 (0.07) | 301.0 | 1.000 |
| Thalamus | 0.935 (0.06) | 0.940 (0.07) | 213.0 | 0.978 |
| Insula | 0.930 (0.04) | 0.930 (0.05) | 214.5 | 0.877 |
| Cingulate gyrus, anterior | 1.005 (0.06) | 1.000 (0.09) | 248.0 | 0.930 |
| Cingulate gyrus, posterior | 1.160 (0.05) | 1.140 (0.07) | 262.0 | 1.000 |
| Cerebellum | 0.960 (0.09) | 0.980 (0.06) | 171.5 | 1.000 |
| Medulla | 0.715 (0.10) | 0.740 (0.09) | 194.0 | 1.000 |
| Midbrain | 0.740 (0.09) | 0.760 (0.06) | 179.0 | 1.000 |
| Pons | 0.625 (0.06) | 0.650 (0.05) | 200.5 | 1.000 |
| Lentiform nucleus | 1.080 (0.07) | 1.100 (0.08) | 203.4 | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vocht, J.; Van Weehaeghe, D.; Ombelet, F.; Masrori, P.; Lamaire, N.; Devrome, M.; Van Esch, H.; Moisse, M.; Koole, M.; Dupont, P.; et al. Differences in Cerebral Glucose Metabolism in ALS Patients with and without C9orf72 and SOD1 Mutations. Cells 2023, 12, 933. https://doi.org/10.3390/cells12060933
De Vocht J, Van Weehaeghe D, Ombelet F, Masrori P, Lamaire N, Devrome M, Van Esch H, Moisse M, Koole M, Dupont P, et al. Differences in Cerebral Glucose Metabolism in ALS Patients with and without C9orf72 and SOD1 Mutations. Cells. 2023; 12(6):933. https://doi.org/10.3390/cells12060933
Chicago/Turabian StyleDe Vocht, Joke, Donatienne Van Weehaeghe, Fouke Ombelet, Pegah Masrori, Nikita Lamaire, Martijn Devrome, Hilde Van Esch, Mathieu Moisse, Michel Koole, Patrick Dupont, and et al. 2023. "Differences in Cerebral Glucose Metabolism in ALS Patients with and without C9orf72 and SOD1 Mutations" Cells 12, no. 6: 933. https://doi.org/10.3390/cells12060933
APA StyleDe Vocht, J., Van Weehaeghe, D., Ombelet, F., Masrori, P., Lamaire, N., Devrome, M., Van Esch, H., Moisse, M., Koole, M., Dupont, P., Van Laere, K., & Van Damme, P. (2023). Differences in Cerebral Glucose Metabolism in ALS Patients with and without C9orf72 and SOD1 Mutations. Cells, 12(6), 933. https://doi.org/10.3390/cells12060933