
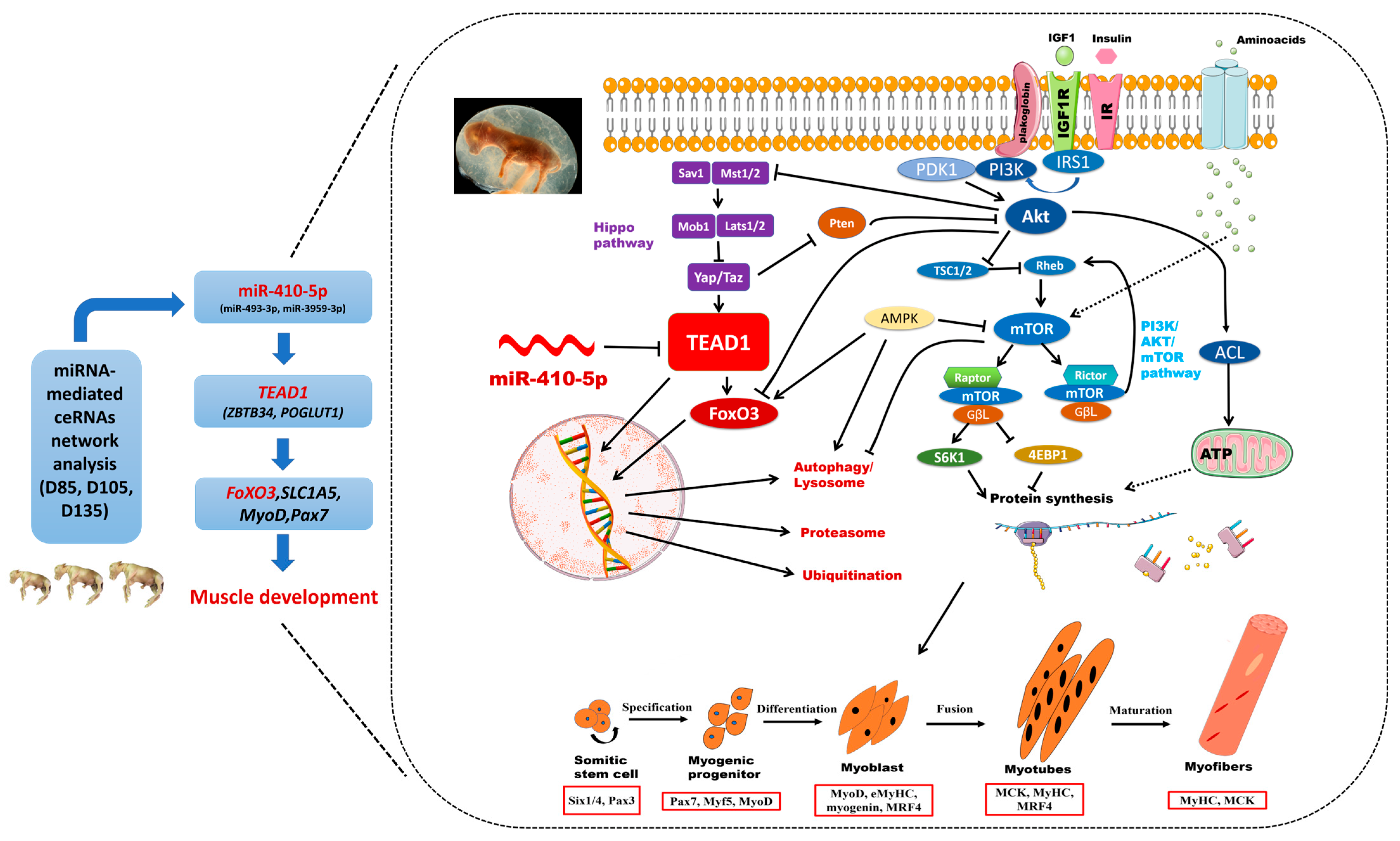
The Molecular Mechanism of the TEAD1 Gene and miR-410-5p Affect Embryonic Skeletal Muscle Development: A miRNA-Mediated ceRNA Network Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequencing and Quantitative Samples
2.2. Small-RNA Sequencing
2.3. Differential Expression and Function Enrichment Analysis of miRNAs
2.4. Construction of the miRNA–mRNA Networks
2.5. Construction of the miRNA–TG–Pathway Network
2.6. Construction of the Integral lncRNA–miRNA–mRNA Interaction Networks
2.7. qRT-PCR of miRNAs and Target Genes in Longissimus Dorsi
2.7.1. RT-PCR of the miRNAs and Target Genes
2.7.2. qRT-PCR of the miRNAs and Target Genes
2.7.3. Statistical Analysis
2.8. Knockdown TEAD1 Gene Expression in Sheep Primary Embryonic Myoblasts
2.9. Dual Luciferase Assay
2.10. Validation of the Targeting Relationship between the DE-miRNAs and TEAD1 Gene
3. Results
3.1. Characterization of the miRNA Expression Profiles
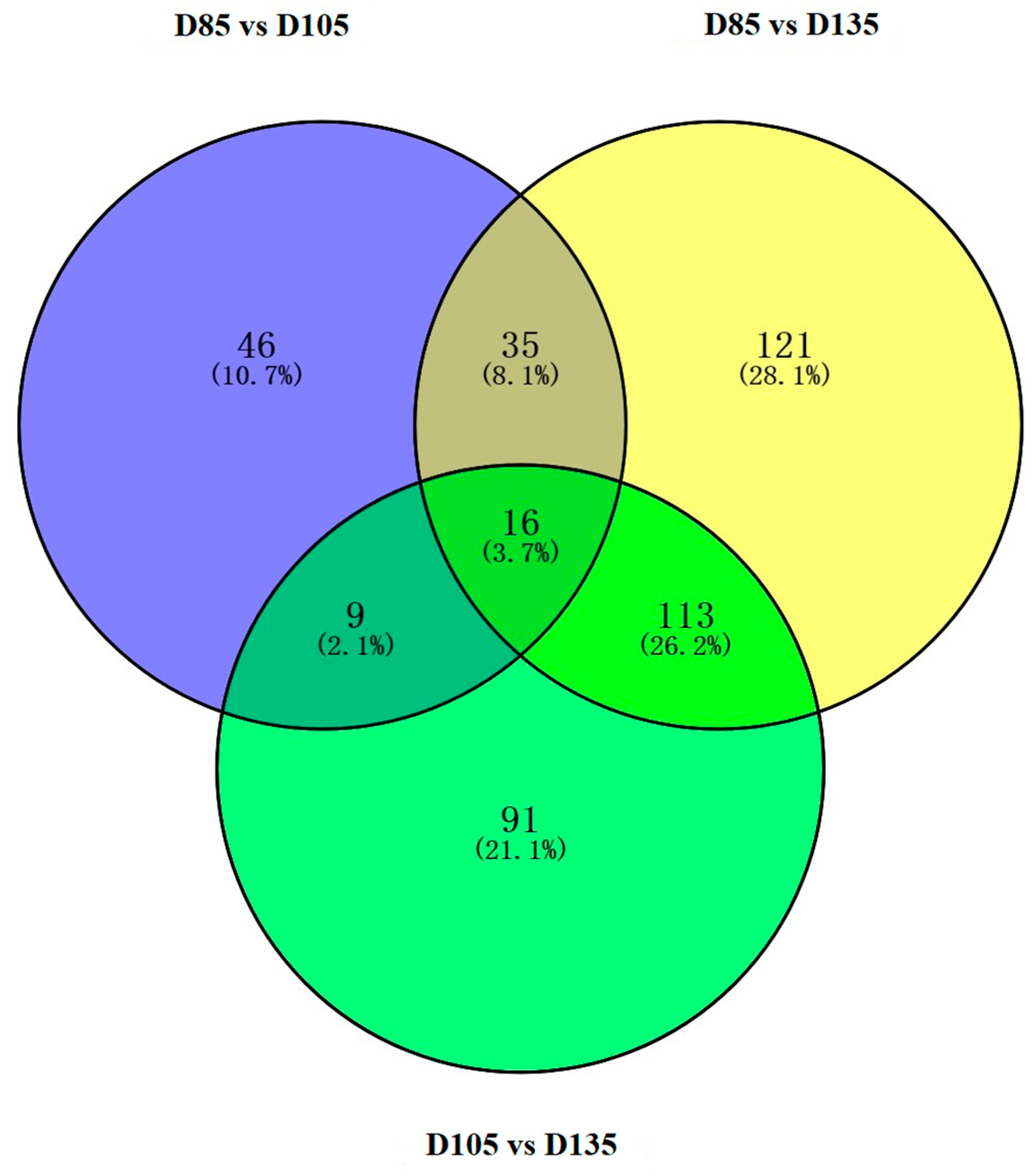
3.2. Identification of the DE-miRNAs and Functional Analysis
3.3. miRNA–mRNA Networks
3.4. miRNA–TG–Pathway Network
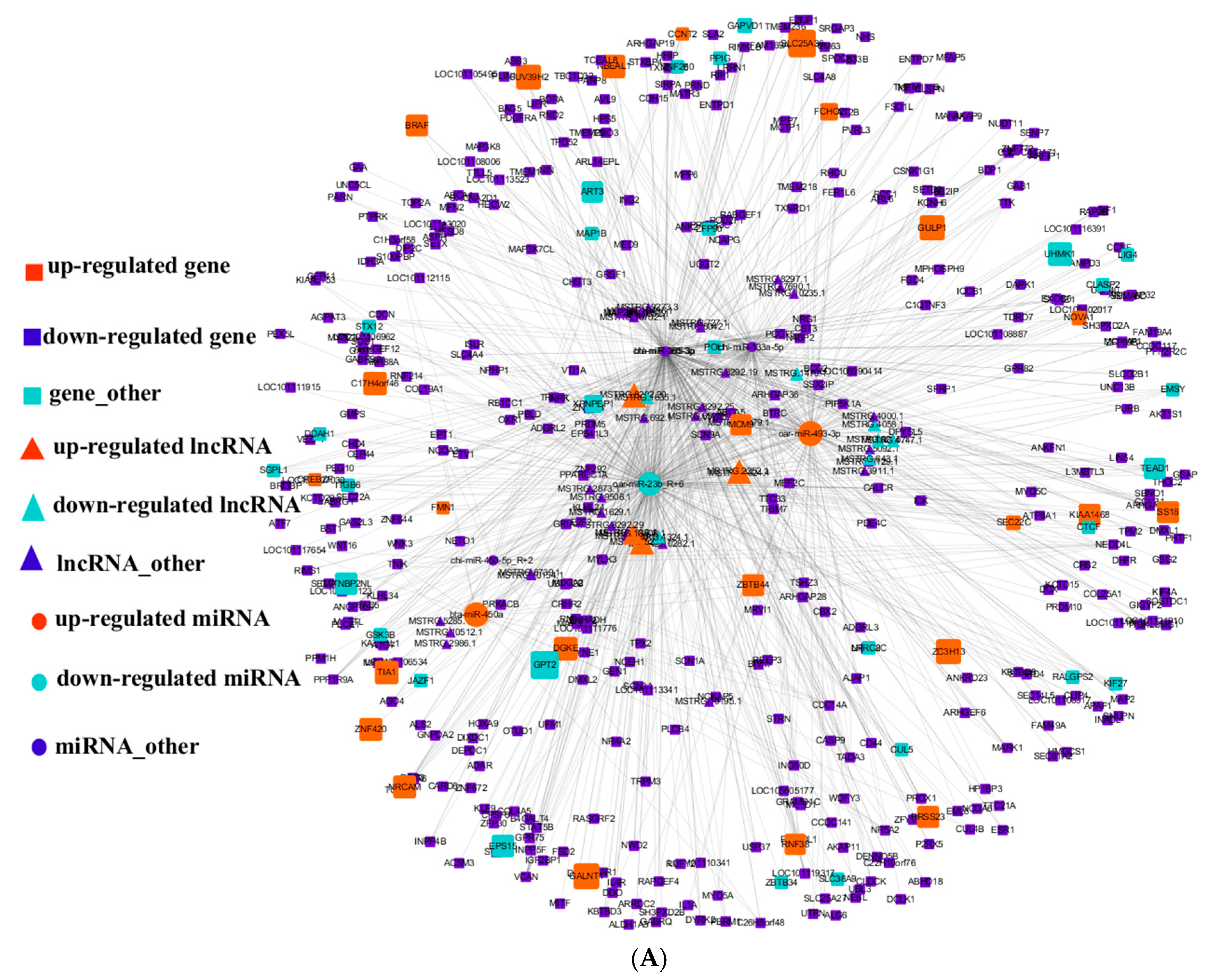
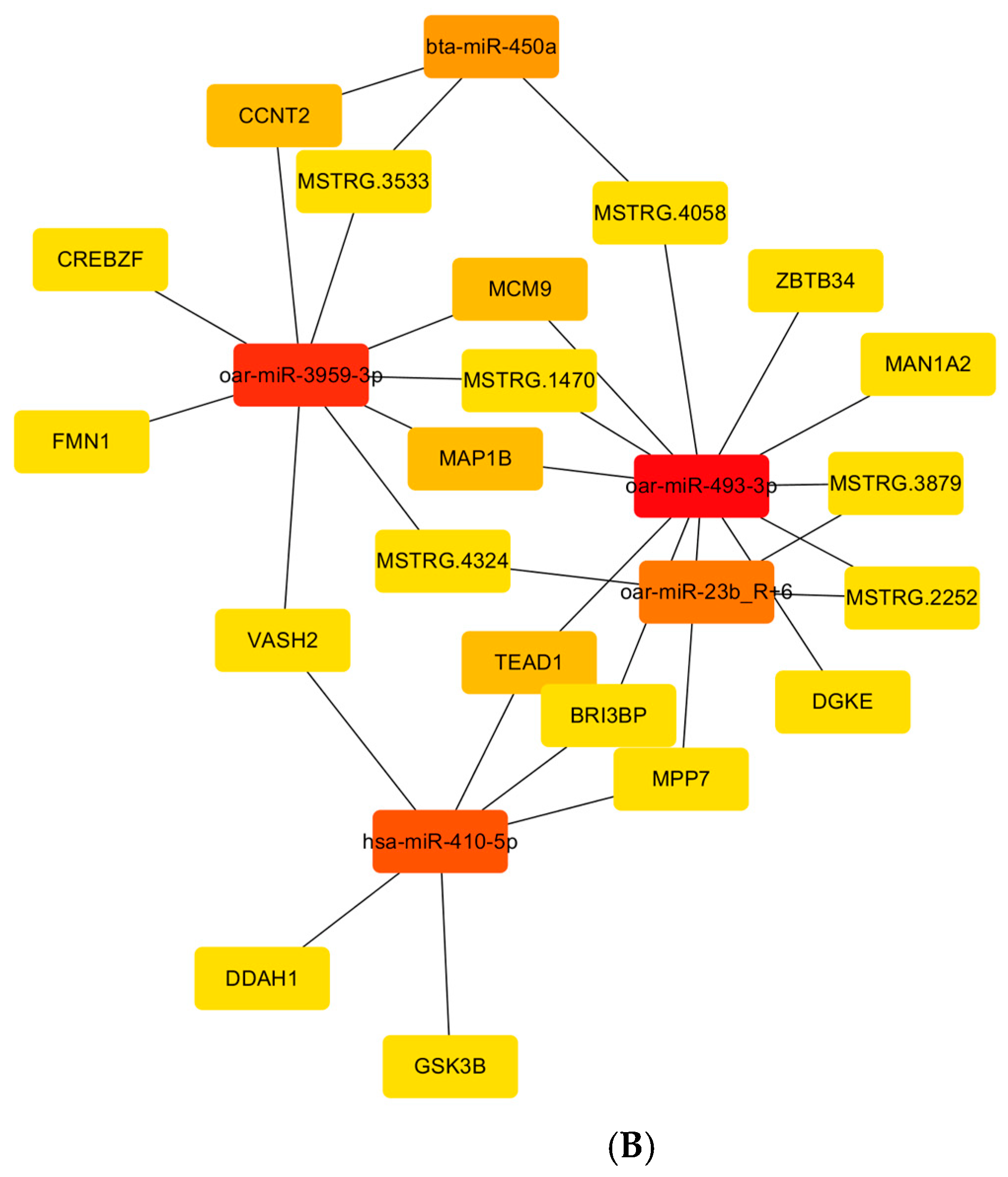
3.5. Integral lncRNA–miRNA–mRNA Interaction Networks
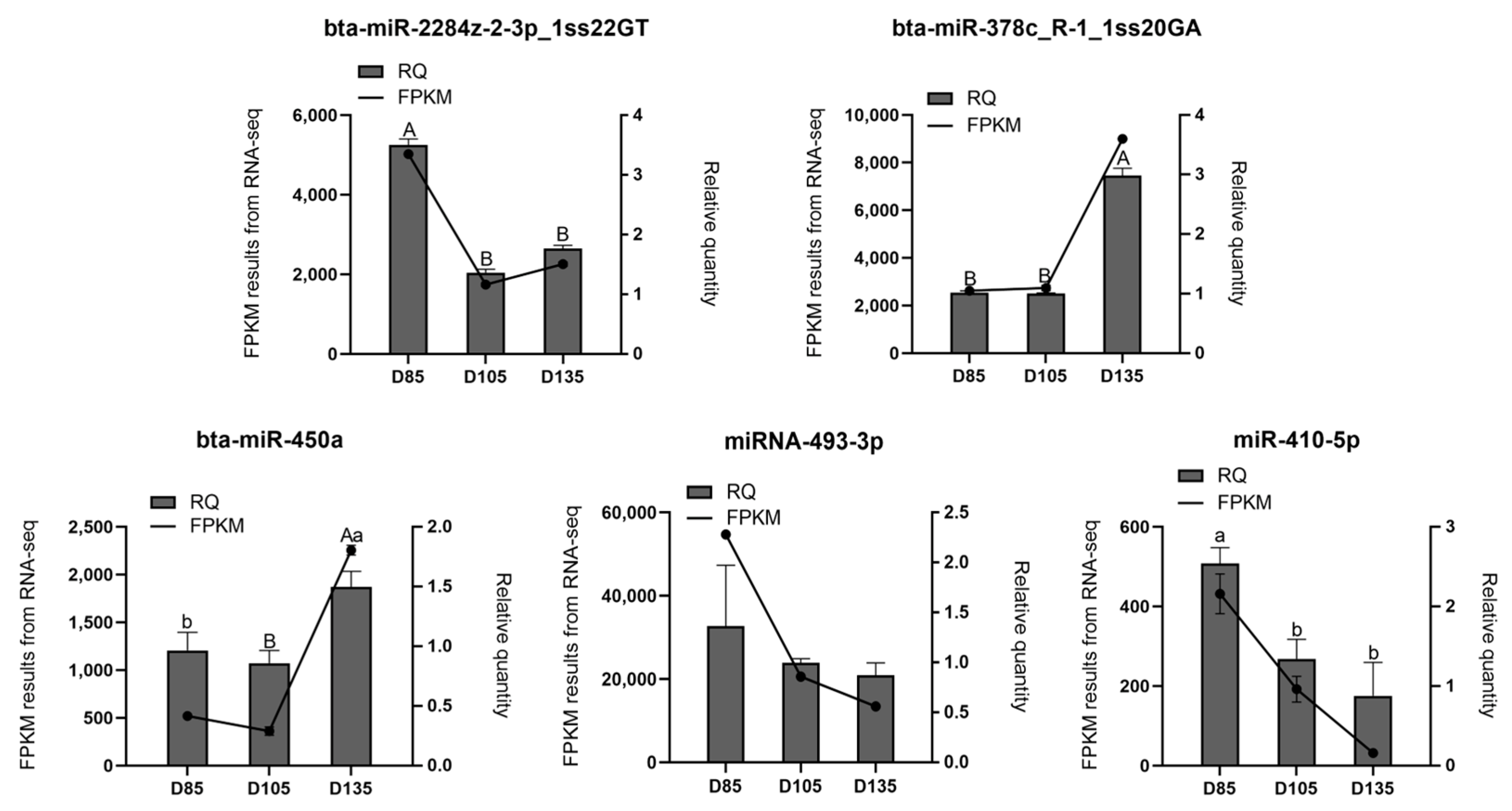
3.6. Validation of the Small-RNA Sequencing Data
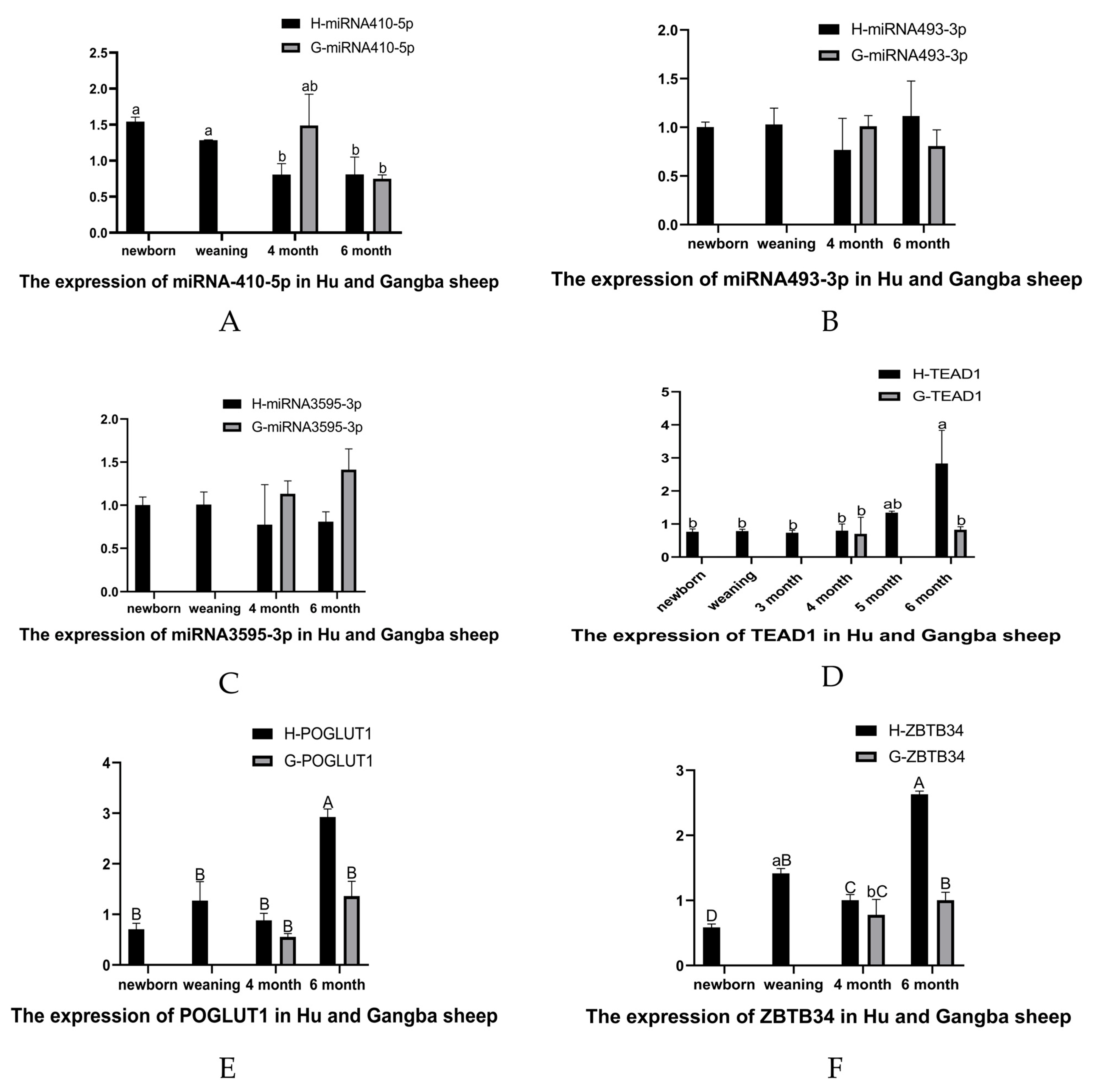
3.7. qRT-PCR of DE-miRNAs and Target Genes in Muscle Tissues of Hu Sheep and Gangba Sheep
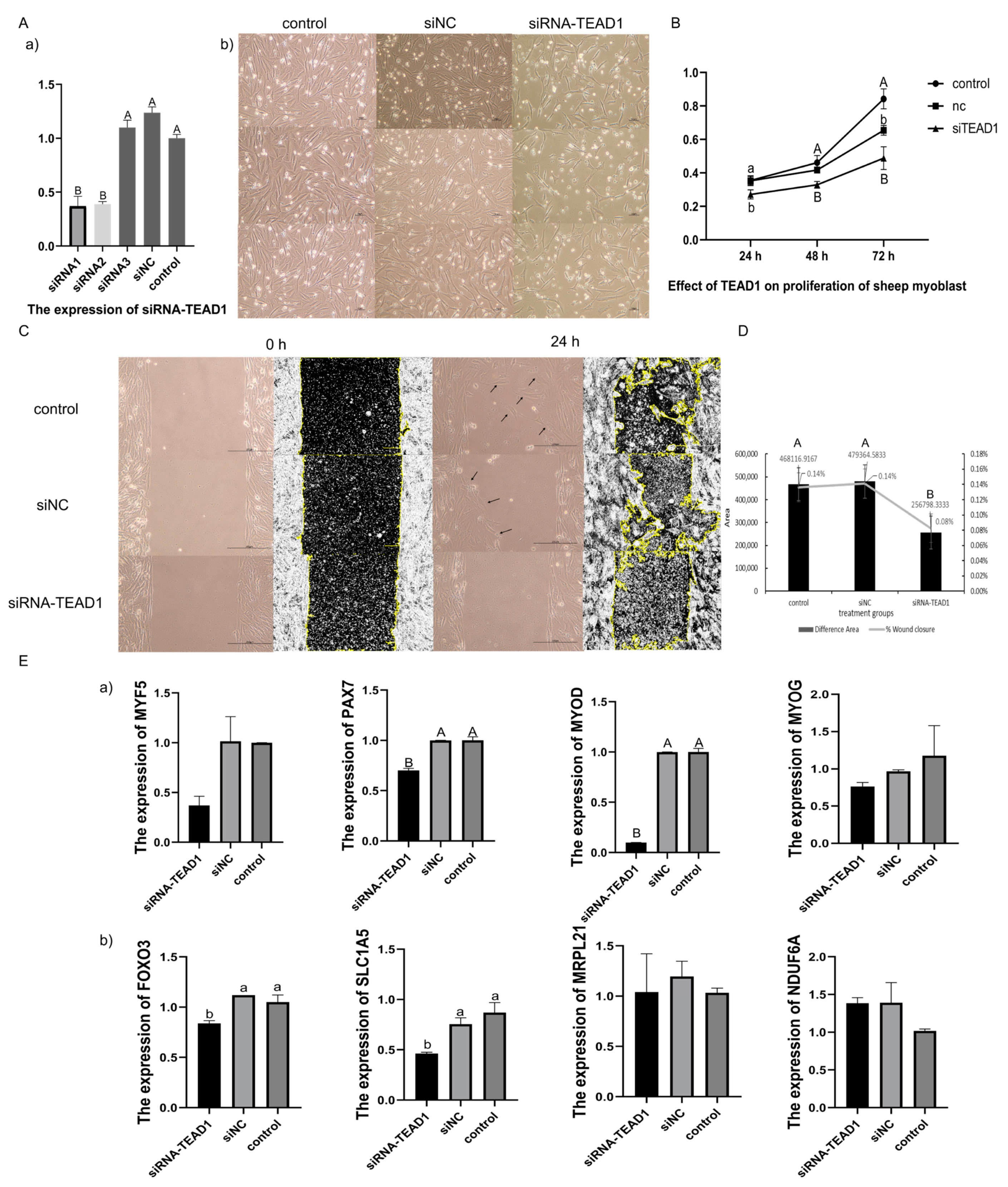
3.8. Knocked-Down TEAD1 Expression Inhibited the Proliferation of Sheep Primary Embryonic Myoblasts
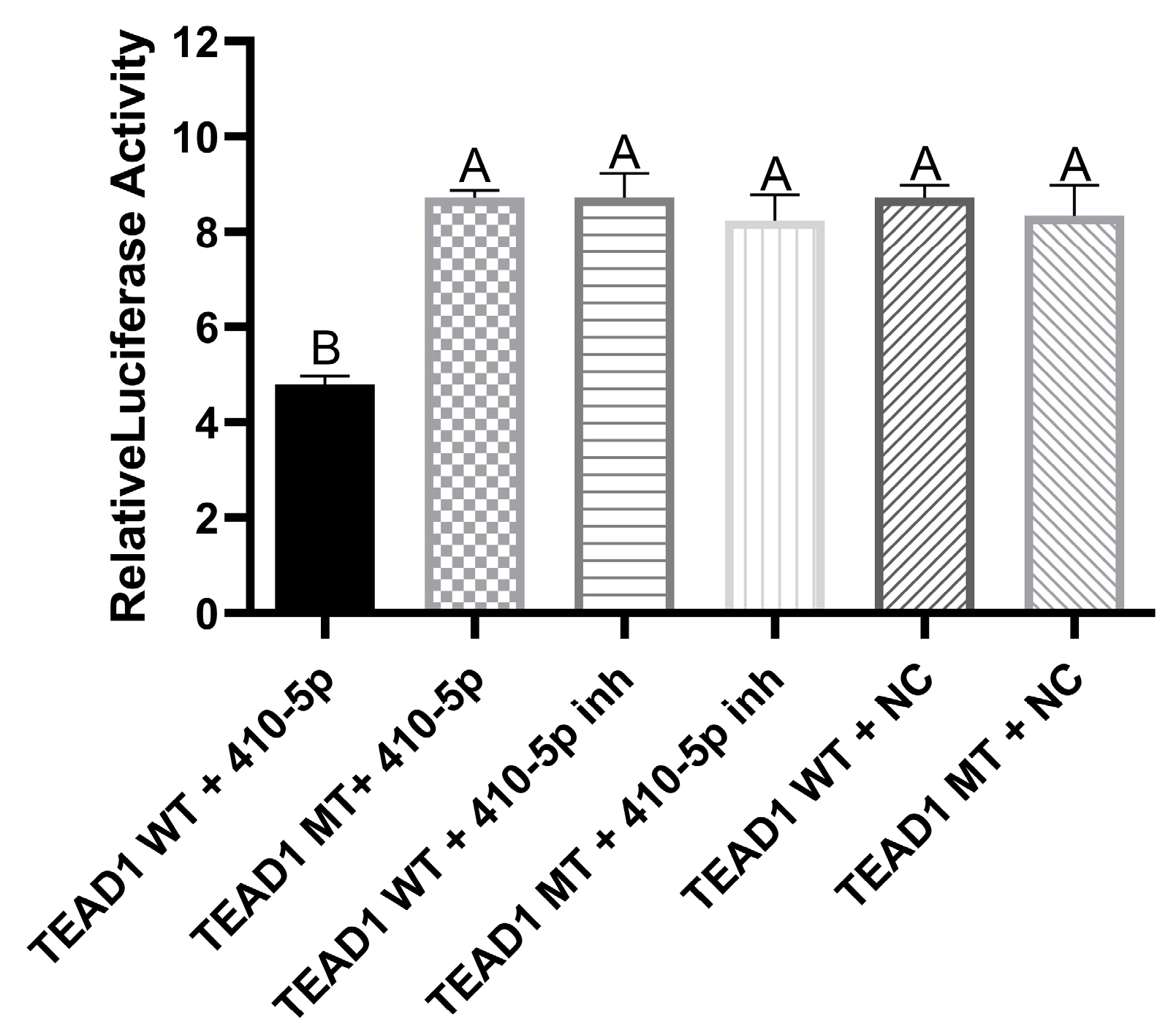
3.9. Validation of the Targeting Relationship between miR-410-5p and TEAD1 Gene by Dual Luciferase Assay
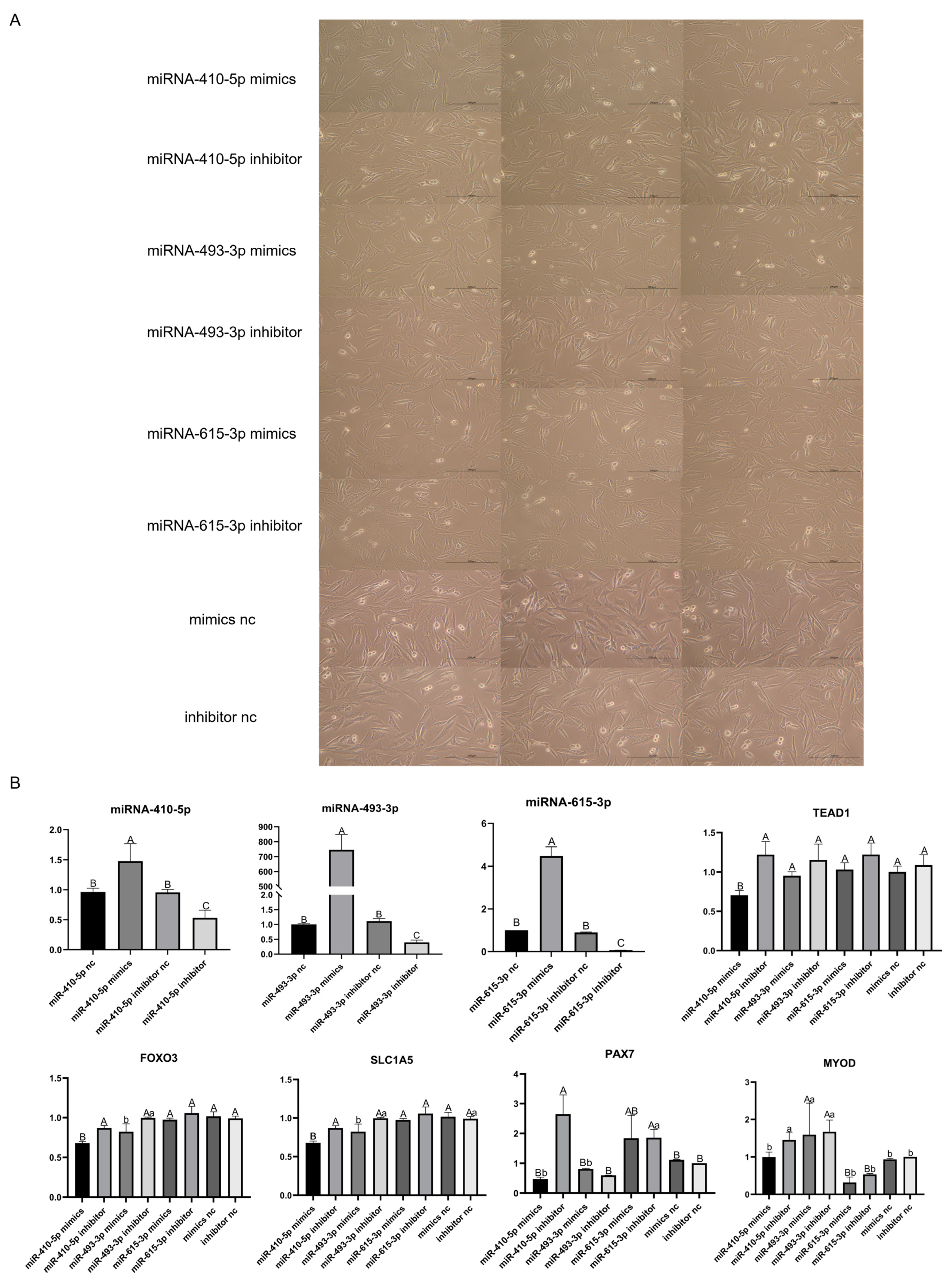
3.10. Validation of the Targeting Relationship between DE-miRNAs and TEAD1 Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehka, L.; Rędowicz, M.J. Mechanisms regulating myoblast fusion: A multilevel interplay. Semin. Cell Dev. Biol. 2020, 104, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Endo, T. Molecular mechanisms of skeletal muscle development, regeneration, and osteogenic conversion. Bone 2015, 80, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Hindi, S.M.; Tajrishi, M.M.; Kumar, A. Signaling Mechanisms in Mammalian Myoblast Fusion. Sci. Signal. 2013, 6, e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building Muscle: Molecular Regulation of Myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, E.A.; Davey, M.G.; Hornick, M.A.; McGovern, P.E.; Mejaddam, A.Y.; Vrecenak, J.D.; Mesas-Burgos, C.; Olive, A.; Caskey, R.C.; Weiland, T.R.; et al. An extra-uterine system to physiologically support the extreme premature lamb. Nat. Commun. 2017, 8, 15112. [Google Scholar] [CrossRef] [Green Version]
- Barry, J.S.; Anthony, R.V. The pregnant sheep as a model for human pregnancy. Theriogenology 2008, 69, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.L. Sheep models of intrauterine growth restriction: Fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008, 35, 730–743. [Google Scholar] [CrossRef]
- Alvites, R.D.; Branquinho, M.V.; Sousa, A.C.; Lopes, B.; Sousa, P.; Mendonca, C.; Atayde, L.M.; Mauricio, A.C. Small ruminants and its use in regenerative medicine: Recent works and future perspectives. Biology 2021, 10, 249. [Google Scholar] [CrossRef]
- McCoard, S.A.; McNabb, W.C.; Peterson, S.W.; McCutcheon, S.N.; Harris, P.M. Muscle growth, cell number, type and morphometry in single and twin fetal lambs during mid to late gestation. Reprod. Fertil. Dev. 2000, 12, 319–327. [Google Scholar] [CrossRef]
- Ren, H.; Li, L.; Su, H.; Xu, L.; Wei, C.; Zhang, L.; Li, H.; Liu, W.; Du, L. Histological and transcriptome-wide level characteristics of fetal myofiber hyperplasia during the second half of gestation in Texel and Ujumqin sheep. BMC Genom. 2011, 12, 411. [Google Scholar] [CrossRef] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Noorbakhsh, J.; Lang, A.H.; Mehta, P. Intrinsic Noise of microRNA-Regulated Genes and the ceRNA Hypothesis. PLoS ONE 2013, 8, e72676. [Google Scholar] [CrossRef]
- Li, X.-Y.; Fu, L.-L.; Cheng, H.-J.; Zhao, S.-H. Advances on microRNA in regulating mammalian skeletal muscle development. Hereditas 2017, 39, 1046–1053. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Stark, A.; Brennecke, J.; Bushati, N.; Russell, R.B.; Cohen, S.M. Animal MicroRNAs Confer Robustness to Gene Expression and Have a Significant Impact on 3′UTR Evolution. Cell 2005, 123, 1133–1146. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, A.; Febrero, M.; Fraiman, R. An anova test for functional data. Comput. Stat. Data Anal. 2004, 47, 111–122. [Google Scholar] [CrossRef]
- Rouder, J.N.; Morey, R.D.; Speckman, P.L.; Province, J.M. Default Bayes factors for ANOVA designs. J. Math. Psychol. 2012, 56, 356–374. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of MicroRNA–Target Recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Siklenka, K.; Arora, S.K.; Ribeiro, P.; Kimmins, S.; Xia, J. miRNet—Dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res. 2016, 44, W135–W141. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xia, J. miRNet-Functional Analysis and Visual Exploration of miRNA-Target Interactions in a Network Context. Methods Mol. Biol. 2018, 1819, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Khorshid, M.; Hausser, J.; Zavolan, M.; van Nimwegen, E. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods 2013, 10, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Shi, T.; Hou, H.; Wang, X.; Zhao, Z.; Shang, M.; Zhang, L. Weighted gene co-expression network analysis for embryo development of skeletal muscle in Chinese Merino Sheep. Acta Vet. Zootech. Sin. 2020, 51, 452–464. [Google Scholar] [CrossRef]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Hu, W.; Shang, M.; Wang, X.; Feng, M.; Liu, Y.; Xiong, J.; Zhang, L. Improvement and characteristic analysis of Hu sheep (Ovis aries) embryo derived myoblasts. Genom. Appl. Biol. 2022, 41, 1440–1451. [Google Scholar] [CrossRef]
- Yang, X.; Zhong, Y.; Wang, D.; Lu, Z. A simple colorimetric method for viable bacteria detection based on cell counting Kit-8. Anal. Methods 2021, 13, 5211–5215. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Fahey, A.J.; Brameld, J.M.; Parr, T.; Buttery, P.J. Ontogeny of factors associated with proliferation and differentiation of muscle in the ovine fetus1,2. J. Anim. Sci. 2005, 83, 2330–2338. [Google Scholar] [CrossRef]
- Ouyang, H.; Wang, Z.; Chen, X.; Yu, J.; Li, Z.; Nie, Q. Proteomic Analysis of Chicken Skeletal Muscle during Embryonic Development. Front. Physiol. 2017, 8, 281. [Google Scholar] [CrossRef] [Green Version]
- Heeley, D.H.; Moir, A.J.; Perry, S.V. Phosphorylation of tropomyosin during development in mammalian striated muscle. FEBS Lett. 1982, 146, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Steelman, C.A.; Recknor, J.C.; Nettleton, D.; Reecy, J.M. Transcriptional profiling of myostatin-knockout mice implicates Wnt signaling in postnatal skeletal muscle growth and hypertrophy. FASEB J. 2006, 20, 580–582. [Google Scholar] [CrossRef]
- Hamelin, M.; Sayd, T.; Chambon, C.; Bouix, J.; Bibeé, B.; Milenkovic, D.; Leveziel, H.; Georges, M.; Clop, A.; Marinova, P.; et al. Proteomic analysis of ovine muscle hypertrophy1. J. Anim. Sci. 2006, 84, 3266–3276. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Zhang, X.; Zhang, H.-K.; Yang, H.-M.; Zhou, Y.-B.; Han, Z.-G. ZBTB34, a novel human BTB/POZ zinc finger protein, is a potential transcriptional repressor. Mol. Cell. Biochem. 2006, 290, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, W.J.; Hong, J.H.; Yaffe, M.B.; Farrance, I.K. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem. J. 2005, 388, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Landin-Malt, A.; Benhaddou, A.; Zider, A.; Flagiello, D. An evolutionary, structural and functional overview of the mammalian TEAD1 and TEAD2 transcription factors. Gene 2016, 591, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Wackerhage, H.; Del, R.D.; Judson, R.N.; Sudol, M.; Sadoshima, J. The Hippo signal transduction network in skeletal and cardiac muscle. Sci. Signal. 2014, 7, e4. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; De Mello, V.; Mohamed, A.; Ortuste, Q.H.; Garcia-Munoz, A.; Al, B.A.; Tremblay, A.M.; von Kriegsheim, A.; Collie-Duguid, E.; Vargesson, N.; et al. Common and Distinctive Functions of the Hippo Effectors Taz and Yap in Skeletal Muscle Stem Cell Function. Stem Cells 2017, 35, 1958–1972. [Google Scholar] [CrossRef] [Green Version]
- Watt, K.I.; Turner, B.J.; Hagg, A.; Zhang, X.; Davey, J.R.; Qian, H.; Beyer, C.; Winbanks, C.E.; Harvey, K.F.; Gregorevic, P. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat. Commun. 2015, 6, 6048. [Google Scholar] [CrossRef] [Green Version]
- Mar, J.H.; Ordahl, C.P. A conserved CATTCCT motif is required for skeletal muscle-specific activity of the cardiac troponin T gene promoter. Proc. Natl. Acad. Sci. USA 1988, 85, 6404–6408. [Google Scholar] [CrossRef] [Green Version]
- Galli, G.G.; Carrara, M.; Yuan, W.-C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo Transducer YAP1 Transforms Activated Satellite Cells and Is a Potent Effector of Embryonal Rhabdomyosarcoma Formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.-S. Transcriptional Co-repressor Function of the Hippo Pathway Transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Watt, K.I.; Judson, R.; Medlow, P.; Reid, K.; Kurth, T.B.; Burniston, J.G.; Ratkevicius, A.; De Bari, C.; Wackerhage, H. Yap is a novel regulator of C2C12 myogenesis. Biochem. Biophys. Res. Commun. 2010, 393, 619–624. [Google Scholar] [CrossRef]
- Judson, R.N.; Tremblay, A.M.; Knopp, P.; White, R.B.; Urcia, R.; De Bari, C.; Zammit, P.S.; Camargo, F.D.; Wackerhage, H. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. J. Cell Sci. 2012, 125, 6009–6019. [Google Scholar] [CrossRef] [Green Version]
- Goodman, C.A.; Dietz, J.M.; Jacobs, B.L.; McNally, R.M.; You, J.-S.; Hornberger, T.A. Yes-Associated Protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett. 2015, 589, 1491–1497. [Google Scholar] [CrossRef] [Green Version]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nature 2012, 14, 1322–1329. [Google Scholar] [CrossRef]
- Cinar, B.; Fang, P.-K.; Lutchman, M.; Di Vizio, D.; Adam, R.M.; Pavlova, N.; Rubin, M.A.; Yelick, P.C.; Freeman, M.R. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 2007, 26, 4523–4534. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.-W.; Yang, S.-J.; Srinivasan, S.; Ye, K. Akt Phosphorylates MstI and Prevents Its Proteolytic Activation, Blocking FOXO3 Phosphorylation and Nuclear Translocation. J. Biol. Chem. 2007, 282, 30836–30844. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T. MCAT Elements and the TEF-1 Family of Transcription Factors in Muscle Development and Disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 8–17. [Google Scholar] [CrossRef]
- Osman, I.; He, X.; Liu, J.; Dong, K.; Wen, T.; Zhang, F.; Yu, L.; Hu, G.; Xin, H.; Zhang, W.; et al. TEAD1 (TEA Domain Transcription Factor 1) Promotes Smooth Muscle Cell Proliferation Through Upregulating SLC1A5 (Solute Carrier Family 1 Member 5)-Mediated Glutamine Uptake. Circ. Res. 2019, 124, 1309–13222. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.; Hu, G.; Wang, Y.; Zhou, J. The Transcription Factor TEAD1 Represses Smooth Muscle-specific Gene Expression by Abolishing Myocardin Function. J. Biol. Chem. 2014, 289, 3308–3316. [Google Scholar] [CrossRef] [Green Version]
- Tsika, R.W.; Schramm, C.; Simmer, G.; Fitzsimons, D.P.; Moss, R.L.; Ji, J. Overexpression of TEAD-1 in Transgenic Mouse Striated Muscles Produces a Slower Skeletal Muscle Contractile Phenotype. J. Biol. Chem. 2008, 283, 36154–36167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsika, R.W.; Ma, L.; Kehat, I.; Schramm, C.; Simmer, G.; Morgan, B.; Fine, D.M.; Hanft, L.M.; McDonald, K.S.; Molkentin, J.D.; et al. TEAD-1 Overexpression in the Mouse Heart Promotes an Age-dependent Heart Dysfunction. J. Biol. Chem. 2010, 285, 13721–13735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, H.; Wu, H.; Qiu, H.; Zeng, C.; Sun, L.; Liu, B. TEAD1 controls C2C12 cell proliferation and differentiation and regulates three novel target genes. Cell. Signal. 2013, 25, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jagannathan, R.; Li, F.; Lee, J.; Balasubramanyam, N.; Kim, B.S.; Yang, P.; Yechoor, V.K.; Moulik, M. Tead1 is required for perinatal cardiomyocyte proliferation. PLoS ONE 2019, 14, e0212017. [Google Scholar] [CrossRef] [Green Version]
- Sawada, A.; Kiyonari, H.; Ukita, K.; Nishioka, N.; Imuta, Y.; Sasaki, H. Redundant Roles of Tead1 and Tead2 in Notochord Development and the Regulation of Cell Proliferation and Survival. Mol. Cell. Biol. 2008, 28, 3177–3189. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Wang, F.; Liu, C.; Xu, X.; Liu, B. TEAD1-dependent expression of the FoxO3a gene in mouse skeletal muscle. BMC Mol. Biol. 2011, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.F.; Larkin, S.B.; Farrance, I.K.; Mar, J.H.; Hall, D.E.; Ordahl, C.P. Muscle-enriched TEF-1 isoforms bind M-CAT elements from muscle-specific promoters and differentially activate transcription. J. Biol. Chem. 1994, 269, 3147–3150. [Google Scholar] [CrossRef]
- Xu, X.; Xing, S.; Du, Z.-Q.; Rothschild, M.F.; Yerle, M.; Liu, B. Porcine TEF1 and RTEF1: Molecular characterization and association analyses with growth traits. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2008, 150, 447–453. [Google Scholar] [CrossRef]
- Xia, X.; Liang, Y.; Zheng, W.; Lin, D.; Sun, S. miR-410-5p promotes the development of diabetic cardiomyopathy by suppressing PIM1-induced anti-apoptosis. Mol. Cell. Probes 2020, 52, 101558. [Google Scholar] [CrossRef]
- Zou, T.; Zhu, M.; Ma, Y.-C.; Xiao, F.; Yu, X.; Xu, L.; Ma, L.-Q.; Yang, J.; Dong, J.-Z. MicroRNA-410-5p exacerbates high-fat diet-induced cardiac remodeling in mice in an endocrine fashion. Sci. Rep. 2018, 8, 8780. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.; Wang, X.; Bi, Y.; Bao, J.; Shang, M.; Zhang, L. The Molecular Mechanism of the TEAD1 Gene and miR-410-5p Affect Embryonic Skeletal Muscle Development: A miRNA-Mediated ceRNA Network Analysis. Cells 2023, 12, 943. https://doi.org/10.3390/cells12060943
Hu W, Wang X, Bi Y, Bao J, Shang M, Zhang L. The Molecular Mechanism of the TEAD1 Gene and miR-410-5p Affect Embryonic Skeletal Muscle Development: A miRNA-Mediated ceRNA Network Analysis. Cells. 2023; 12(6):943. https://doi.org/10.3390/cells12060943
Chicago/Turabian StyleHu, Wenping, Xinyue Wang, Yazhen Bi, Jingjing Bao, Mingyu Shang, and Li Zhang. 2023. "The Molecular Mechanism of the TEAD1 Gene and miR-410-5p Affect Embryonic Skeletal Muscle Development: A miRNA-Mediated ceRNA Network Analysis" Cells 12, no. 6: 943. https://doi.org/10.3390/cells12060943
APA StyleHu, W., Wang, X., Bi, Y., Bao, J., Shang, M., & Zhang, L. (2023). The Molecular Mechanism of the TEAD1 Gene and miR-410-5p Affect Embryonic Skeletal Muscle Development: A miRNA-Mediated ceRNA Network Analysis. Cells, 12(6), 943. https://doi.org/10.3390/cells12060943