
Targeting ARID1A-Deficient Cancers: An Immune-Metabolic Perspective

 , ,
, ,  and
and
Abstract
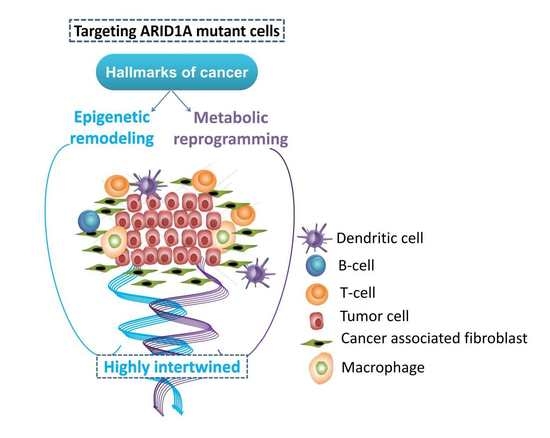
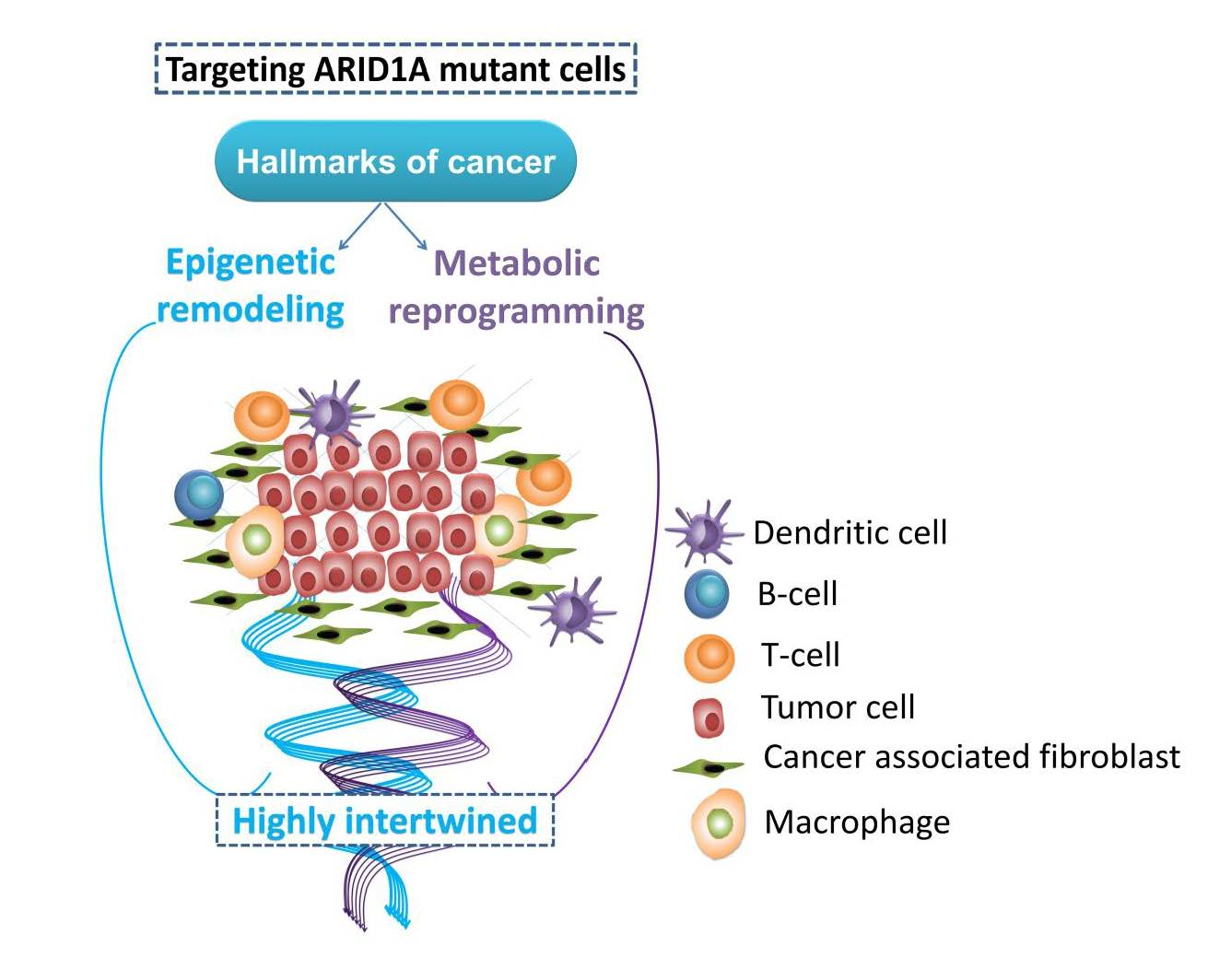
:
1. Introduction
2. ARID1A: Context-Dependent Functions
3. ARID1A: Synthetic Lethality and Beyond
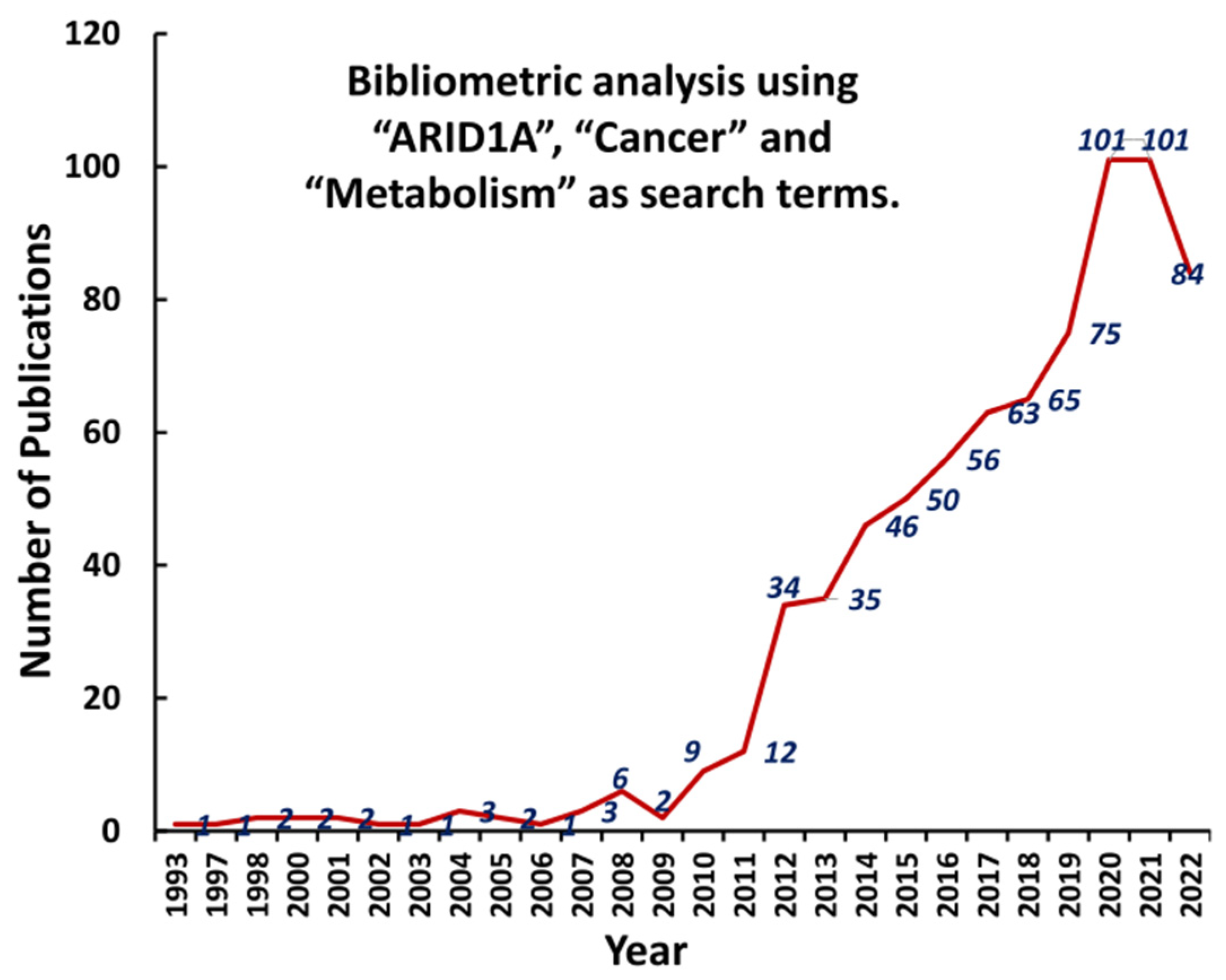
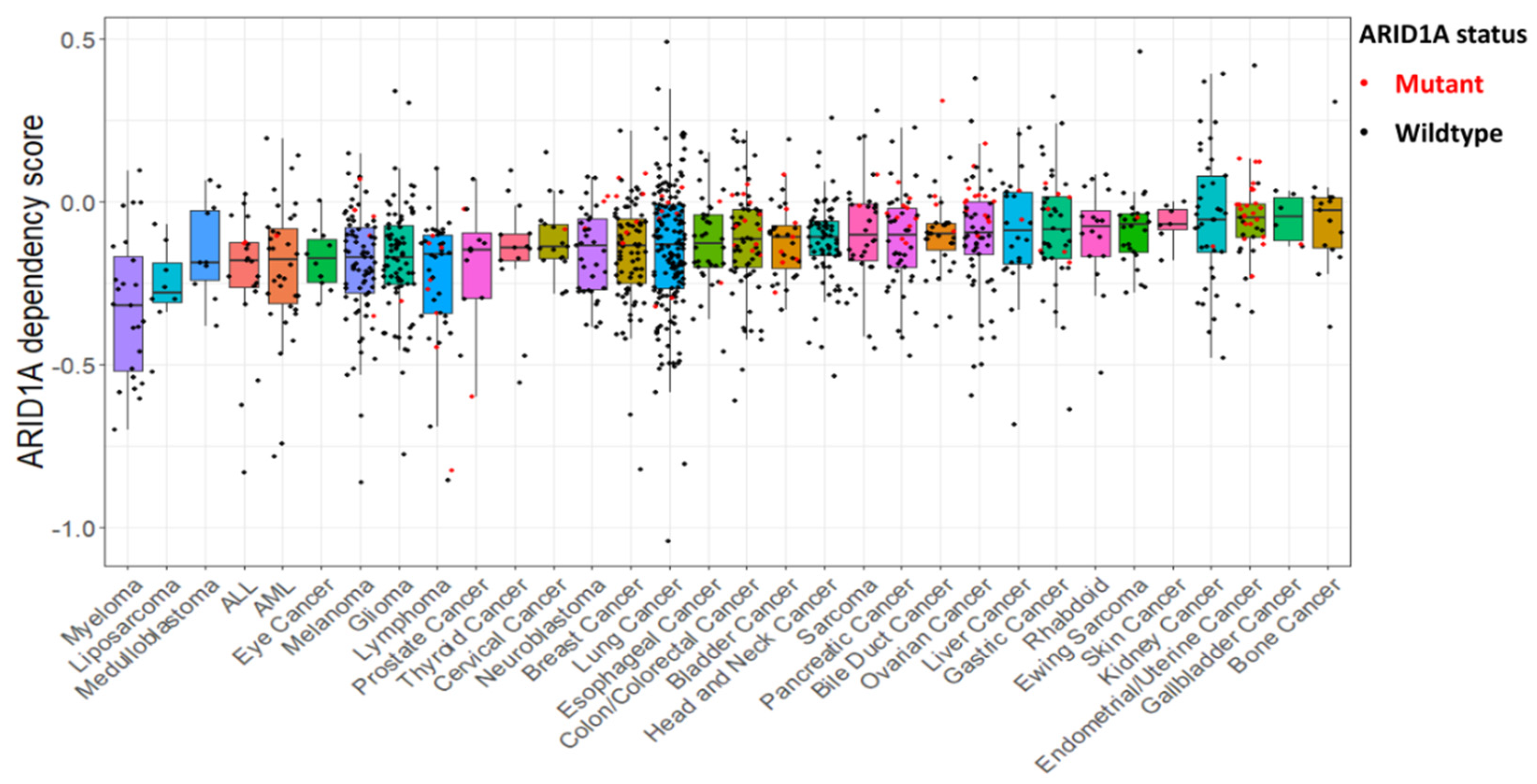
4. ARID1A-Deficiency: An Immune-Metabolic Vulnerability
4.1. Metabolic Rewiring in ARID1A-Deficient Cancers
4.2. ARID1A as a Modulator of Antitumor Immunity
5. Future Outlooks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, M.; Zhang, J.; Huang, W.; Wang, Y. Interplay Among Metabolism, Epigenetic Modifications, and Gene Expression in Cancer. Front. Cell Dev. Biol. 2021, 9, 793428. [Google Scholar] [CrossRef]
- van Weverwijk, A.; de Visser, K.E. Mechanisms driving the immunoregulatory function of cancer cells. Nat. Rev. Cancer 2023. [CrossRef] [PubMed]
- Akbari, B.; Hosseini, Z.; Shahabinejad, P.; Ghassemi, S.; Mirzaei, H.R.; O’Connor, R.S. Metabolic and epigenetic orchestration of (CAR) T cell fate and function. Cancer Lett. 2022, 550, 215948. [Google Scholar] [CrossRef]
- Ferrara, A.L.; Liotti, A.; Pezone, A.; De Rosa, V. Therapeutic opportunities to modulate immune tolerance through the metabolism-chromatin axis. Trends Endo Metab. 2022, 33, 507–521. [Google Scholar] [CrossRef]
- Belk, J.A.; Daniel, B.; Satpathy, A.T. Epigenetic regulation of T cell exhaustion. Nat. Immunol. 2022, 23, 848–860. [Google Scholar] [CrossRef]
- Møller, S.H.; Hsueh, P.C.; Yu, Y.R.; Zhang, L.; Ho, P.C. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab. 2022, 34, 378–395. [Google Scholar] [CrossRef]
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 2021, 20, 171. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Scharping, N.E.; Goldrath, A.W. CD8+ T cell metabolism in infection and cancer. Nat. Rev. Immunol. 2021, 21, 718–738. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.; Kato, S.; Sicklick, J.K.; Kurzrock, R. Targeting ARID1A mutations in cancer. Cancer Treat. Rev. 2021, 100, 102287. [Google Scholar] [CrossRef] [PubMed]
- Mandal, J.; Mandal, P.; Wang, T.L.; Shih, I.M. Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response. J. Biomed. Sci. 2022, 29, 71. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tang, C. The Role of ARID1A in Tumors: Tumor Initiation or Tumor Suppression? Front. Oncol. 2021, 11, 745187. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih, I.e.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [Green Version]
- McConechy, M.K.; Ding, J.; Cheang, M.C.; Wiegand, K.C.; Senz, J.; Tone, A.A.; Yang, W.; Prentice, L.M.; Tse, K.; Zeng, T.; et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol. 2012, 228, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Xu, Y.; Wu, W.; Wang, P.; Wang, Y.; Jiang, H.; Zhu, J. ARID1A variations in cholangiocarcinoma: Clinical significances and molecular mechanisms. Front. Oncol. 2021, 11, 693295. [Google Scholar] [CrossRef]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, W.; Jiang, Z.; Tang, F.; Ding, L.; Xu, W.; Ruan, L. Roles of ARID1A variations in colorectal cancer: A collaborative review. Mol. Med. 2022, 28, 1–16. [Google Scholar] [CrossRef]
- Lissanu Deribe, Y.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Sausen, M.; Leary, R.J.; Jones, S.; Wu, J.; Reynolds, C.P.; Liu, X.; Blackford, A.; Parmigiani, G.; Diaz, L.A., Jr.; Papadopoulos, N.; et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013, 45, 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Tate, P.; Hu, P.; Tjian, R.; Skarnes, W.C.; Wang, Z. ES Cell Pluripotency and Germ-Layer Formation Require the SWI/SNF Chromatin Remodeling Component BAF250a. Proc. Natl. Acad. Sci. USA 2008, 105, 6656–6661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Zhang, M.; Ji, F.; Zhao, J.; Wang, Y.; Wang, W.; Zhang, S.; Ma, H.; Wang, Y.; Jiao, J. Microglia homeostasis mediated by epigenetic ARID1A regulates neural progenitor cells response and leads to autism-like behaviors. Mol. Psychiatry 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Shi, R.; Liu, Y.; Ke, J.; Liu, X.; Du, H.Z.; Liu, C.M. Abnormal microglial polarization induced by Arid1a deletion leads to neuronal differentiation deficits. Cell Prolif. 2022, 55, e13314. [Google Scholar] [CrossRef]
- Li, W.; Yang, L.; He, Q.; Hu, C.; Zhu, L.; Ma, X.; Ma, X.; Bao, S.; Li, L.; Chen, Y.; et al. A Homeostatic Arid1a-Dependent Permissive Chromatin State Licenses Hepatocyte Responsiveness to Liver-Injury-Associated YAP Signaling. Cell Stem Cell. 2019, 25, 54–68.e5. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Lei, L.; Su, Y.; Zou, Y.; Liu, H.; Jiao, A.; Zhang, C.; Liu, J.; Li, W.; et al. Arid1a promotes thymocyte development through β-selection-dependent and β-selection-independent mechanisms. Immunology 2022, 165, 402–413. [Google Scholar] [CrossRef]
- Han, L.; Madan, V.; Mayakonda, A.; Dakle, P.; Woon, T.W.; Shyamsunder, P.; Nordin, H.B.M.; Cao, Z.; Sundaresan, J.; Lei, I.; et al. Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia 2019, 33, 2291–2305. [Google Scholar] [CrossRef] [Green Version]
- Mendiratta, G.; Ke, E.; Aziz, M.; Liarakos, D.; Tong, M.; Stites, E.C. Cancer gene mutation frequencies for the U.S. population. Nat. Commun. 2021, 12, 5961. [Google Scholar] [CrossRef] [PubMed]
- Martin-Romano, P.; Colmet-Daage, L.; Morel, D.; Baldini, C.; Verlingue, L.; Bahleda, R.; Gazzah, A.; Champiat, S.; Varga, A.; Michot, J.M.; et al. Epigenetic gene alterations in metastatic solid tumours: Results from the prospective precision medicine MOSCATO and MATCH-R trials. Eur. J. Cancer 2022, 173, 133–145. [Google Scholar] [CrossRef]
- Burkhardt, B.; Michgehl, U.; Rohde, J.; Erdmann, T.; Berning, P.; Reutter, K.; Rohde, M.; Borkhardt, A.; Burmeister, T.; Dave, S.; et al. Clinical relevance of molecular characteristics in Burkitt lymphoma differs according to age. Nat. Commun. 2022, 13, 3881. [Google Scholar] [CrossRef]
- Waks, A.G.; Kim, D.; Jain, E.; Snow, C.; Kirkner, G.J.; Rosenberg, S.M.; Oh, C.; Poorvu, P.D.; Ruddy, K.J.; Tamimi, R.M.; et al. Somatic and Germline Genomic Alterations in Very Young Women with Breast Cancer. Clin. Cancer Res. 2022, 28, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.N.; Roberts, C.W. ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [Green Version]
- McDonald, E.R., 3rd; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Lebedev, T.; Buzdin, A.; Khabusheva, E.; Spirin, P.; Suntsova, M.; Sorokin, M.; Popenko, V.; Rubtsov, P.; Prassolov, V. Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling. Int. J. Mol. Sci. 2022, 23, 7724. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Lin, H.; Jin, R.; Liu, J.; Liu, X.; Meng, N.; Cai, X. The Clinicopathologic Significance of BAF250a (ARID1A) Expression in Hepatocellular Carcinoma. Pathol. Oncol. Res. 2016, 22, 453–459. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.-C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 32, 574–589. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W.J.; Hoivik, E.A.; Halle, M.K.; Taylor-Weiner, A.; Cherniack, A.D.; Berg, A.; Holst, F.; Zack, T.; Werner, H.; Staby, K.; et al. The Genomic Landscape and Evolution of Endometrial Carcinoma Progression and Abdominopelvic Metastasis. Nat. Genet. 2016, 48, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Dobzhansky, T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946, 31, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliaroli, L.; Porazzi, P.; Curtis, A.T.; Scopa, C.; Mikkers, H.M.M.; Freund, C.; Daxinger, L.; Deliard, S.; Welsh, S.A.; Offley, S.; et al. Inability to switch from ARID1A-BAF to ARID1B-BAF impairs exit from pluripotency and commitment towards neural crest formation in ARID1B-related neurodevelopmental disorders. Nat. Commun. 2021, 12, 6469. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, D.; Saw, P.E.; Song, E. Turning cold tumors hot: From molecular mechanisms to clinical applications. Trends. Immunol. 2022, 43, 523–545. [Google Scholar] [CrossRef]
- Kirchhammer, N.; Trefny, M.P.; Auf der Maur, P.; Läubli, H.; Zippelius, A. Combination cancer immunotherapies: Emerging treatment strategies adapted to the tumor microenvironment. Sci. Transl. Med. 2022, 14, eabo3605. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Dis. 2020, 19, 776–800. [Google Scholar] [CrossRef]
- Villanueva, L.; Álvarez-Errico, D.; Esteller, M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell. 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [Green Version]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Fukumoto, T.; Lin, J.; Nacarelli, T.; Wang, Y.; Ong, D.; Liu, H.; Fatkhutdinov, N.; Zundell, J.A.; Karakashev, S.; et al. Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat. Cancer 2021, 2, 189–200. [Google Scholar] [CrossRef]
- Phase Arend, R. 1 Trial of CB-839 in Combination With Niraparib in Platinum Resistant BRCA-Wild-Type Ovarian Cancer Patients [Internet]. clinicaltrials.gov; 2021. May. Report No.: NCT03944902. Available online: https://clinicaltrials.gov/ct2/show/NCT03944902 (accessed on 31 December 2022).
- Clemente, V.; Hoshino, A.; Shetty, M.; Nelson, A.; Erickson, B.K.; Baker, R.; Rubin, N.; Khalifa, M.; Weroha, S.J.; Lou, E.; et al. GLS1 is a protective factor in patients with ovarian clear cell carcinoma and its expression does not correlate with ARID1A-mutated tumors. Cancer Res. Commun. 2022, 2, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Zhang, F.K.; Ni, Q.Z.; Wang, K.; Cao, H.J.; Guan, D.X.; Zhang, E.B.; Ma, N.; Wang, Y.K.; Zheng, Q.W.; Xu, S.; et al. Targeting USP9X-AMPK Axis in ARID1A-Deficient Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 101–127. [Google Scholar] [CrossRef] [PubMed]
- Penugurti, V.; Mishra, Y.G.; Manavathi, B. AMPK: An odyssey of a metabolic regulator, a tumor suppressor, and now a contextual oncogene. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188785. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tay, N.S.C.; Jaynes, P.; Anbuselvan, A.; Ramachandran, G.K.; Wardyn, J.D.; Hoppe, M.; Hoang, P.M.; Peng, Y.; Lim, S.; et al. PLK1 inhibition selectively induces apoptosis in ARID1A deficient cells through uncoupling of oxygen consumption from ATP production. Oncogene 2022, 41, 1986–2002. [Google Scholar] [CrossRef]
- Shakeel, I.; Basheer, N.; Hasan, G.M.; Afzal, M.; Hassan, M.I. Polo-like kinase 1 as an emerging drug target: Structure, function and therapeutic implications. J. Drug Target. 2021, 29, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Sun, S.; Xu, H.; Zhao, Z.; Han, Z.; Jia, J.; Wu, D.; Lu, J.; Liu, H.; Yu, R.; et al. Combined delivery of temozolomide and siPLK1 using targeted nanoparticles to enhance temozolomide sensitivity in glioma. Int. J. Nanomed. 2020, 15, 3347–3362. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Wang, Z.; Liu, F.; Zhang, L.; Ke, J.; Xu, X.; Zhang, Y.; Yuan, Y.; Wei, T.; et al. Chromatin Remodeling Induced by ARID1A Loss in Lung Cancer Promotes Glycolysis and Confers JQ1 Vulnerability. Cancer Res. 2022, 82, 791–804. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef] [Green Version]
- Mehrvarz Sarshekeh, A.; Alshenaifi, J.; Roszik, J.; Manyam, G.C.; Advani, S.M.; Katkhuda, R.; Verma, A.; Lam, M.; Willis, J.; Shen, J.P.; et al. ARID1A Mutation May Define an Immunologically Active Subgroup in Patients with Microsatellite Stable Colorectal Cancer. Clin. Cancer Res. 2021, 27, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Buglioni, S.; Melucci, E.; Sperati, F.; Pallocca, M.; Terrenato, I.; De Nicola, F.; Goeman, F.; Casini, B.; Amoreo, C.A.; Gallo, E.; et al. The clinical significance of PD-L1 in advanced gastric cancer is dependent on ARID1A mutations and ATM expression. Oncoimmunology 2018, 7, e1457602. [Google Scholar] [CrossRef] [Green Version]
- Botta, G.P.; Kato, S.; Patel, H.; Fanta, P.; Lee, S.; Okamura, R.; Kurzrock, R. SWI/SNF complex alterations as a biomarker of immunotherapy efficacy in pancreatic cancer. JCI Insight 2021, 6, e150453. [Google Scholar] [CrossRef]
- Li, J.; Wang, W.; Zhang, Y.; Cieślik, M.; Guo, J.; Tan, M.; Green, M.D.; Wang, W.; Lin, H.; Li, W.; et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J. Clin. Investig. 2020, 130, 2712–2726. [Google Scholar] [CrossRef]
- Fukumoto, T.; Fatkhutdinov, N.; Zundell, J.A.; Tcyganov, E.N.; Nacarelli, T.; Karakashev, S.; Wu, S.; Liu, Q.; Gabrilovich, D.I.; Zhang, R. HDAC6 Inhibition Synergizes with Anti-PD-L1 Therapy in ARID1A-Inactivated Ovarian Cancer. Cancer Res. 2019, 79, 5482–5489. [Google Scholar] [CrossRef]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J., III; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.; Chen, Y.; Anandhan, S.; Szabo, P.M.; Basu, S.; Blando, J.M.; Liu, W.; Zhang, J.; Natarajan, S.M.; Xiong, L.; et al. ARID1A mutation plus CXCL13 expression act as combinatorial biomarkers to predict responses to immune checkpoint therapy in mUCC. Sci. Transl. Med. 2020, 12, eabc4220. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.e.M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Chui, M.H.; Suryo Rahmanto, Y.; Yu, Z.C.; Shamanna, R.A.; Bellani, M.A.; Gaillard, S.; Ayhan, A.; Viswanathan, A.; Seidman, M.M.; et al. Loss of ARID1A in Tumor Cells Renders Selective Vulnerability to Combined Ionizing Radiation and PARP Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5584–5594. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef] [PubMed]
- Belk, J.A.; Yao, W.; Ly, N.; Freitas, K.A.; Chen, Y.T.; Shi, Q.; Valencia, A.M.; Shifrut, E.; Kale, N.; Yost, K.E.; et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 2022, 40, 768–786.e7. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.; Ray, J.; de Boer, C.; Jenkins, R.; Lieb, D.; Chen, J.; Frederick, D.; Barzily-Rokni, M.; et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2018, 175, 998–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Liu, Q.; Han, Y.; Pei, S.; Cheng, B.; Xu, J.; Miao, X.; Pan, Q.; Wang, H.; Guo, J.; et al. ARID1A loss induces polymorphonuclear myeloid-derived suppressor cell chemotaxis and promotes prostate cancer progression. Nat. Commun. 2022, 13, 7281. [Google Scholar] [CrossRef]
- Arner, E.N.; Rathmell, J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
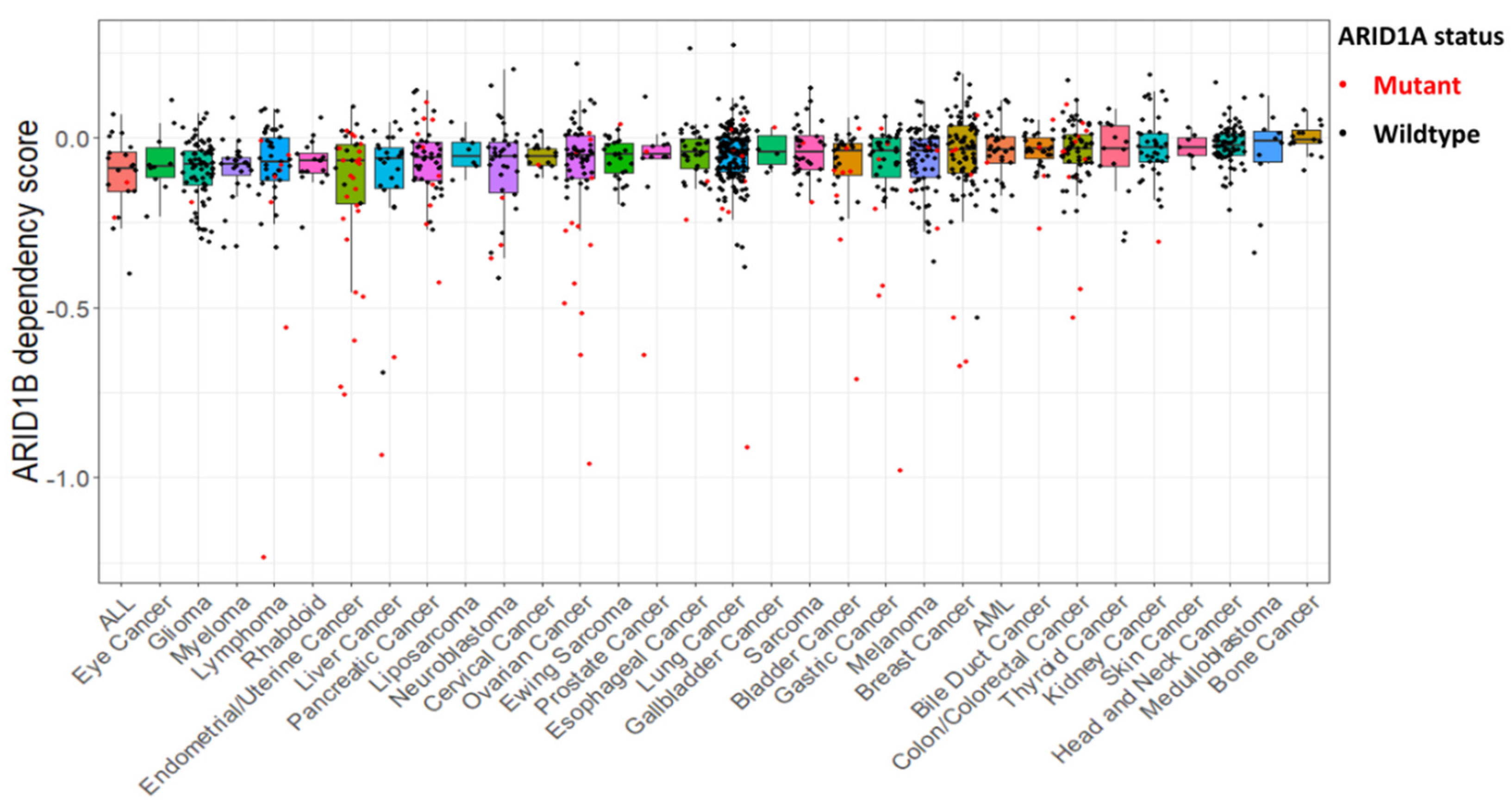{kind=link}
| Types of Cancer | Mutation Rate | Reference |
|---|---|---|
| Ovarian clear cell carcinoma | 40–57% | [15] |
| High-grade endometrioid adenocarcinoma | 60% | [16] |
| Low-grade endometrioid adenocarcinoma | 47% | [16] |
| Ovarian endometrioid carcinomas | 30% | [15] |
| Cholangiocarcinomas, intrahepatic | 7–36% | [17] |
| Cholangiocarcinomas, extrahepatic | 5–12% | [17] |
| Gastric adenocarcinomas | 18–27% | [18] |
| Hepatocellular carcinoma | 10–17% | [19] |
| Pancreatic ductal adenocarcinoma | 6–10% | [20] |
| Colon cancer | 10% | [21] |
| Lung cancer | 8% | [22] |
| Neuroblastoma | 6% | [23] |
| Title | Cancer/Stage | Drug/Combination | Phases | Status | NCT# |
|---|---|---|---|---|---|
| Phase II Study of Tazemetostat in Solid Tumors Harboring an ARID1A Mutation | Solid Tumor|ARID1A Gene Mutation | Drug: Tazemetostat | Phase 2 | Recruiting | NCT05023655 |
| PD-1 Combined with Dasatinib for as Third-line Treatment for ARID1A Mutation Advanced NSCLC | NSCLC Stage IV|ARID1A|PD-1 | Drug: PD-1 plus Dasatinib | Phase 2 | Unknown status | NCT04284202 |
| Bevacizumab and/or Niraparib in Patients with Recurrent Endometrial and/or Ovarian Cancer With ARID1A Mutation | Recurrent Endometrial Carcinoma|Recurrent Ovarian Carcinoma|ARID1A Gene Mutation | Drug: Bevacizumab|Drug: Niraparib | Phase 2 | Not yet recruiting | NCT05523440 |
| ATr Inhibitor in Combination with Olaparib in Gynaecological Cancers With ARId1A Loss or no Loss | Gynaecological Cancers | Drug: AZD6738|Drug: Olaparib | Phase 2 | Recruiting | NCT04065269 |
| Nivolumab for the Treatment of Metastatic or Unresectable Solid Tumors with ARID1A Mutation and CXCL13 Expression | Metastatic Malignant Solid Neoplasm|Unresectable Solid Neoplasm | Biological: Nivolumab | Phase 2 | Recruiting | NCT04957615 |
| Nivolumab for the Treatment of Patients with Metastatic Urothelial Cancer With ARID1A Mutation and Stratify Response Based on CXCL13 Expression | Multiple cancers | Other: Diagnostic Laboratory Biomarker Analysis|Biological: Nivolumab | Phase 2 | Not yet recruiting | NCT04953104 |
| Evaluating Safety & Efficacy Belinostat Combo w Nivo Alone & w Ipi in Patients w Treated Metastatic/Advanced Carcinomas w ARID1A Lof Mutation | Metastatic Adenocarcinoma | Drug: Belinostat|Drug: nivolumab|Drug: ipilimumab | Phase 1 | Withdrawn | NCT04315155 |
| Tremelimumab, Durvalumab, and Belinostat for the Treatment of ARID1A Mutated Metastatic or Unresectable, Locally Advanced Urothelial Carcinoma | Multiple cancers | Drug: Belinostat|Biological: Durvalumab|Biological: Tremelimumab | Phase 1 | Recruiting | NCT05154994 |
| PLX2853 as a Single Agent in Advanced Gynecological Malignancies and in Combination with Carboplatin in Platinum-Resistant Epithelial Ovarian Cancer | Gynecologic Neoplasms|Epithelial Ovarian Cancer | Drug: PLX2853|Drug: Carboplatin | Phase 1|Phase 2 | Terminated | NCT04493619 |
| Dasatinib in Treating Patients with Recurrent or Persistent Ovarian, Fallopian Tube, Endometrial or Peritoneal Cancer | Multiple cancers | Drug: Dasatinib|Other: Laboratory Biomarker Analysis | Phase 2 | Active, not recruiting | NCT02059265 |
| JAB-2485 Activity in Adult Patients with Advanced Solid Tumors | Multiple cancers | Drug: JAB-2485 (Aurora A inhibitor) | Phase 1|Phase 2 | Not yet recruiting | NCT05490472 |
| Olaparib in Treating Patients with Metastatic Biliary Tract Cancer With Aberrant DNA Repair Gene Mutations | Multiple cancers | Drug: Olaparib | Phase 2 | Recruiting | NCT04042831 |
| Prognostic Biomarkers in Patients with Urothelial Carcinoma | Bladder Cancer | Completed | NCT04872036 | ||
| A Study of PLX2853 in Advanced Malignancies. | Multiple cancers | Drug: PLX2853 | Phase 1 | Completed | NCT03297424 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebedev, T.; Kousar, R.; Patrick, B.; Usama, M.; Lee, M.-K.; Tan, M.; Li, X.-G. Targeting ARID1A-Deficient Cancers: An Immune-Metabolic Perspective. Cells 2023, 12, 952. https://doi.org/10.3390/cells12060952
Lebedev T, Kousar R, Patrick B, Usama M, Lee M-K, Tan M, Li X-G. Targeting ARID1A-Deficient Cancers: An Immune-Metabolic Perspective. Cells. 2023; 12(6):952. https://doi.org/10.3390/cells12060952
Chicago/Turabian StyleLebedev, Timofey, Rubina Kousar, Bbumba Patrick, Muhammad Usama, Meng-Kuei Lee, Ming Tan, and Xing-Guo Li. 2023. "Targeting ARID1A-Deficient Cancers: An Immune-Metabolic Perspective" Cells 12, no. 6: 952. https://doi.org/10.3390/cells12060952
APA StyleLebedev, T., Kousar, R., Patrick, B., Usama, M., Lee, M. -K., Tan, M., & Li, X. -G. (2023). Targeting ARID1A-Deficient Cancers: An Immune-Metabolic Perspective. Cells, 12(6), 952. https://doi.org/10.3390/cells12060952