
High Glucose Activates Prolyl Hydroxylases and Disrupts HIF-α Signaling via the P53/TIGAR Pathway in Cardiomyocyte

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Experimental Mice
2.2. Cell Culture
2.3. Western Blot Analysis
2.4. RNA Interference
2.5. Adenoviral Vectors Transfection in H9c2 Cell Lines
2.6. Metabolic Assays
2.7. Immunofluorescence Analysis
2.8. Statistical Analysis
3. Results
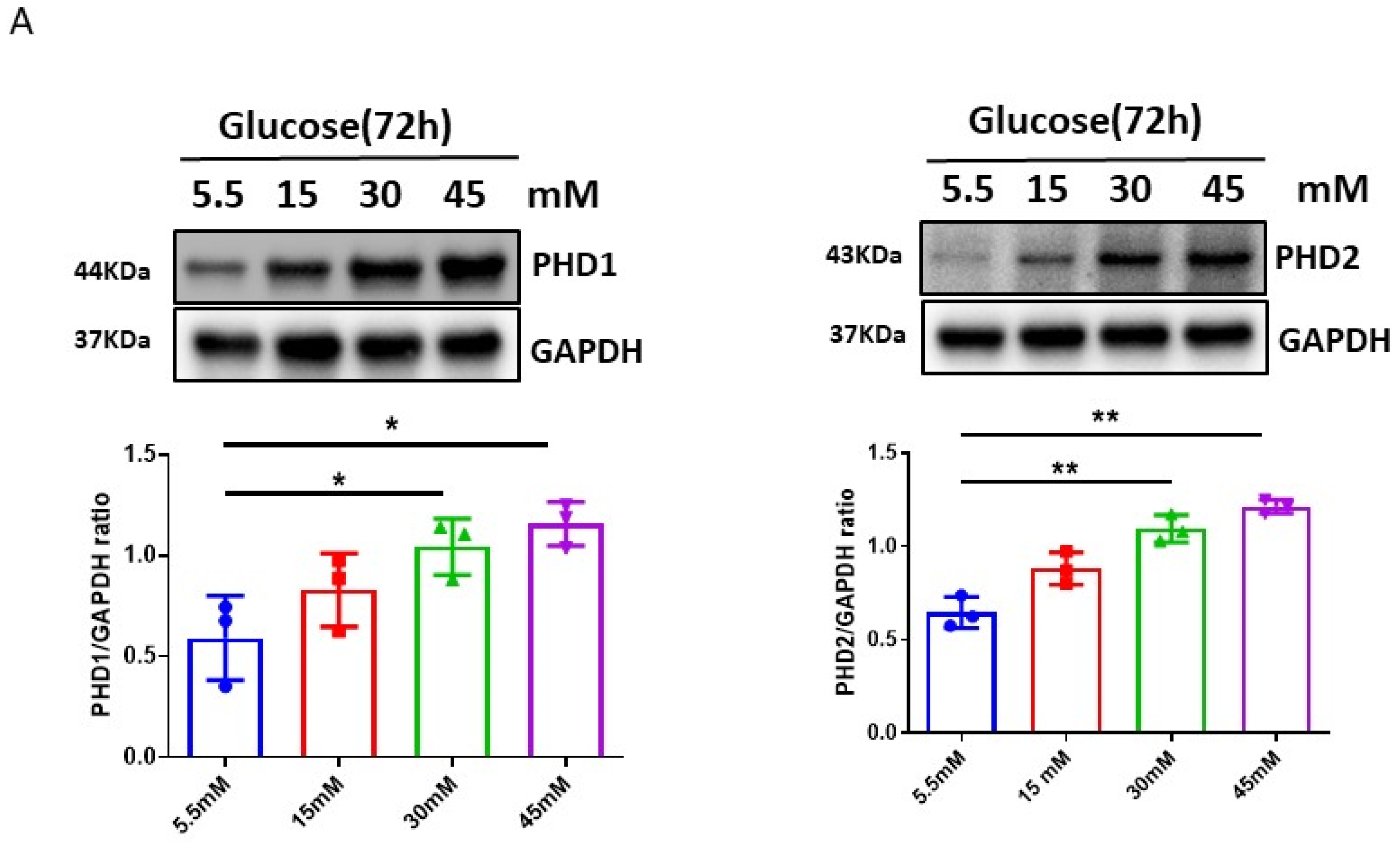
3.1. High Glucose (HG) Upregulated PHDs and Reduced HIF-α Expression in H9c2 Cells
3.2. Pretreatment with p53 siRNA Reduced Expression of PHDs and Improved HIF-α Signaling in H9c2 Cells under HG Conditions
3.3. Pretreatment with p53 siRNA Altered H9c2 Cell Metabolism under HG Conditions
3.4. Pretreatment with TIGAR siRNA Reduced PHDs and Enhanced HIF-α Signaling in H9c2 Cells
3.5. SIRT3 Regulates PHDs-HIF-α Signaling Pathway in H9c2 Cells and in Diabetic Hearts
4. Discussion
5. Conclusions
5.1. Novelty and Significance
5.1.1. What Is New?
- High glucose upregulated PHDs, thus resulting in an impairment of hypoxic signaling and an increased oxygen consumption rate (OCR) in H9c2 cells, which may contribute to diabetic cardiomyopathy.
- High glucose upregulated p53 and TIGAR expression in cardiomyocytes. Pretreatment with p53 and TIGAR siRNA reversed the impairment of hypoxic signaling and decreased oxygen consumption rate (OCR) in cardiomyocytes.
- SIRT3 suppressed p53 and blunted diabetes- and hyperglycemia-induced disruption of PHDs-HIF-α signaling.
5.1.2. What Is Relevant?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DCM | Diabetic cardiomyopathy |
| HG | High glucose |
| HIF-1α | Hypoxia-inducible factor-1α |
| HIF-2α | Hypoxia-inducible factor-2α |
| PHD1 | Prolyl hydroxylase-1 |
| PHD2 | Prolyl hydroxylase-2 |
| P53 | Tumor suppression factor p53 |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| VEGF | Vascular endothelial growth factor |
| Ang-1 | Angiopoietin 1 |
| TGF-β | Tissue growth factor-β |
| FSP-1 | Fibroblast specific protein-1 |
| SIRT3 | Sirtuin 3 |
| ECAR | Extracellular acidification rate |
| OCR | Oxygen Consumption Rate |
References
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2017, 61, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, M.; Manrique, A.; Cariou, B.; Prieur, X. Diabetes-related cardiomyopathy: The sweet story of glucose overload from epidemiology to cellular pathways. Diabetes Metab. 2018, 45, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.M.; Waqar, T.; Howarth, F.C.; Adeghate, E.; Bidasee, K.; Singh, J. Hyperglycemia-induced cardiac contractile dysfunction in the diabetic heart. Heart Fail. Rev. 2017, 23, 37–54. [Google Scholar] [CrossRef]
- Di Filippo, C.; Marfella, R.; Cuzzocrea, S.; Piegari, E.; Petronella, P.; Giugliano, D.; Rossi, F.; D’Amico, M. Hyper-glycemia in streptozotocin-induced diabetic rat increases infarct size associated with low levels of myocardial HO-1 during ischemia/reperfusion. Diabetes 2005, 54, 803–810. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.; Calvillo, L.; Tillmanns, J.; Elbing, I.; Dienesch, C.; Bischoff, H.; Ertl, G.; Bauersachs, J. Repetitive post-prandial hyperglycemia increases cardiac ischemia/reperfusion injury: Prevention by the alpha-glucosidase inhibitor acarbose. FASEB J. 2005, 19, 591–593. [Google Scholar] [CrossRef]
- Lehto, S.; Pyörälä, K.; Miettinen, H.; Rönnemaa, T.; Palomäki, P.; Tuomilehto, J.; Laakso, M. Myocardial infarct size and mortality in patients with non-insulin-dependent diabetes mellitus. J. Intern. Med. 1994, 236, 291–297. [Google Scholar] [CrossRef]
- Liu, X.; Wei, J.; Peng, D.H.; Layne, M.D.; Yet, S.F. Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes 2005, 5, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Marfella, R.; Di Filippo, C.; Esposito, K.; Nappo, F.; Piegari, E.; Cuzzocrea, S.; Berrino, L.; Rossi, F.; Giugliano, D.; D’Amico, M. Absence of inducible nitric oxide synthase reduces myocardial damage during ischemia reperfusion in strepto-zotocin-induced hyperglycemic mice. Diabetes 2004, 53, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Morrish, N.J.; WHO Multinational Study Group; Wang, S.-L.; Stevens, L.K.; Fuller, J.H.; Keen, H. Mortality and causes of death in the WHO multinational study of vascular disease in diabetes. Diabetologia 2001, 44, S14–S21. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Maitland, A.; Weisel, R.D.; Li, S.-H.; Fedak, P.W.; Pomroy, N.C.; Mickle, D.A.; Li, R.-K.; Ko, L.; Rao, V. Hyperglycemia exaggerates ischemia-reperfusion–induced cardiomyocyte injury: Reversal with endothelin antagonism. J. Thorac. Cardiovasc. Surg. 2002, 123, 1120–1124. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, M.; Inoue, I.; Kawagoe, T.; Shimatani, Y.; Kurisu, S.; Nishioka, K.; Umemura, T.; Nakamura, S.; Yoshida, M. Effect of acute hyperglycemia on the ischemic preconditioning effect of prodromal angina pectoris in patients with a first anterior wall acute myocardial infarction. Am. J. Cardiol. 2003, 92, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Kersten, J.R.; Schmeling, T.J.; Orth, K.G.; Pagel, P.S.; Warltier, D.C. Acute hyperglycemia abolishes ischemic preconditioning in vivo. Am. J. Physiol. 1998, 275, H721–H725. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, K.; Ito, H.; Ikushima, M.; Kawano, S.; Okamura, A.; Asano, K.; Kuroda, T.; Tanaka, K.; Masuyama, T.; Hori, M.; et al. Association between hyperglycemia and the no-reflow phenomenon in patients with acute myocardial infarction. J. Am. Coll. Cardiol. 2003, 41, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragonés, J.; Fraisl, P.; Baes, M.; Carmeliet, P. Oxygen Sensors at the Crossroad of Metabolism. Cell Metab. 2009, 9, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragonés, J.; Schneider, M.; Van Geyte, K.; Fraisl, P.; Dresselaers, T.; Mazzone, M.; Dirkx, R.; Zacchigna, S.; Lemieux, H.; Jeoung, N.H.; et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat. Genet. 2008, 40, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Fraisl, P.; Aragonés, J.; Carmeliet, P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 2009, 8, 139–152. [Google Scholar] [CrossRef]
- Schneider, M.; Van Geyte, K.; Fraisl, P.; Kiss, J.; Aragonés, J.; Mazzone, M.; Mairbäurl, H.; De Bock, K.; Jeoung, N.H.; Mollenhauer, M.; et al. Loss or Silencing of the PHD1 Prolyl Hydroxylase Protects Livers of Mice Against Ischemia/Reperfusion Injury. Gastroenterology 2010, 138, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Heather, L.C.; Clarke, K. Metabolism, hypoxia and the diabetic heart. J. Mol. Cell. Cardiol. 2011, 50, 598–605. [Google Scholar] [CrossRef]
- Marfella, R.; D’Amico, M.; Di Filippo, C.; Piegari, E.; Nappo, F.; Esposito, K.; Berrino, L.; Rossi, F.; Giugliano, D. Myocardial infarction in diabetic rats: Role of hyperglycaemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia 2002, 45, 1172–1181. [Google Scholar]
- Marfella, R.; Esposito, K.; Nappo, F.; Siniscalchi, M.; Sasso, F.C.; Portoghese, M.; Di Marino, M.P.; Baldi, A.; Cuzzocrea, S.; Di Filippo, C.; et al. Expression of Angiogenic Factors During Acute Coronary Syndromes in Human Type 2 Diabetes. Diabetes 2004, 53, 2383–2391. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, A.R.; Toscano, A.; Pereira, P.; Girao, H.; Goncalves, L.; Marques, C. Hyperglycemia-induced degradation of HIF-1alpha contributes to impaired response of cardiomyocytes to hypoxia. Rev. Port. Cardiol. 2017, 36, 367–373. [Google Scholar] [CrossRef]
- Chen, J.X.; Stinnett, A. Ang-1 gene therapy inhibits hypoxia-inducible factor-1alpha (HIF-1alpha)-prolyl-4-hydroxylase-2, stabilizes HIF-1alpha expression, and normalizes immature vasculature in db/db mice. Diabetes 2008, 57, 3335–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.X.; Stinnett, A. Disruption of Ang-1/Tie-2 signaling contributes to the impaired myocardial vascular maturation and angiogenesis in type II diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.-H.; Zeng, H.; Stinnett, A.; Yu, H.; Aschner, J.L.; Liao, D.-F.; Chen, J.-X. Critical role of angiopoietins/Tie-2 in hyperglycemic exacerbation of myocardial infarction and impaired angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2547–H2557. [Google Scholar] [CrossRef] [Green Version]
- Birks, E.J.; Latif, N.; Enesa, K.; Folkvang, T.; Luong le, A.; Sarathchandra, P.; Khan, M.; Ovaa, H.; Terracciano, C.M.; Barton, P.J.; et al. Elevated p53 expression is associated with dysregulation of the ubiqui-tin-proteasome system in dilated cardiomyopathy. Cardiovasc. Res. 2008, 79, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Conte, J.V., Jr.; Foster, A.H.; McLaughlin, J.S.; Wei, C. Increased p53 protein expression in human failing myocardium. J. Heart Lung Transplant. 1999, 18, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Morita, H.; Komuro, I. Novel molecular mechanisms and regeneration therapy for heart failure. J. Mol. Cell. Cardiol. 2016, 92, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced in-hibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Nithipongvanitch, R.; Ittarat, W.; Velez, J.M.; Zhao, R.; Clair, D.K.S.; Oberley, T.D. Evidence for p53 as Guardian of the Cardiomyocyte Mitochondrial Genome Following Acute Adriamycin Treatment. J. Histochem. Cytochem. 2007, 55, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Shizukuda, Y.; Matoba, S.; Mian, O.Y.; Nguyen, T.; Hwang, P.M. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol. Cell Biochem. 2005, 273, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Gogiraju, R.; Xu, X.; Bochenek, M.L.; Steinbrecher, J.H.; Lehnart, S.E.; Wenzel, P.; Kessel, M.; Zeisberg, E.M.; Dobbelstein, M.; Schäfer, K. Endothelial p53 Deletion Improves Angiogenesis and Prevents Cardiac Fibrosis and Heart Failure Induced by Pressure Overload in Mice. J. Am. Heart Assoc. 2015, 4, 1770. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Bolanos, J.P. TIGAR’s promiscuity. Biochem. J. 2014, 458, e5–e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Matoba, S.; Iwai-Kanai, E.; Nakamura, H.; Kimata, M.; Nakaoka, M.; Katamura, M.; Okawa, Y.; Ariyoshi, M.; Mita, Y.; et al. p53-TIGAR axis attenuates mitophagy to ex-acerbate cardiac damage after ischemia. J. Mol. Cell Cardiol. 2012, 52, 175–184. [Google Scholar] [CrossRef]
- Kimata, M.; Matoba, S.; Iwai-Kanai, E.; Nakamura, H.; Hoshino, A.; Nakaoka, M.; Katamura, M.; Okawa, Y.; Mita, Y.; Okigaki, M.; et al. p53 and TIGAR regulate cardiac myocyte energy homeostasis under hypoxic stress. Am. J. Physiol. Circ. Physiol. 2010, 299, H1908–H1916. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zeng, H.; He, X.; Chen, J. Sirtuin 3 Alleviates Diabetic Cardiomyopathy by Regulating TIGAR and Cardiomyocyte Metabolism. J. Am. Heart Assoc. 2021, 10, e018913. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5’ enhancer. Circ. Res. 1995, 77, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia-induced activation of HIF-1: Role of HIF-1α-Hsp90 interaction. FEBS Lett. 1999, 460, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively ex-pressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-inducible Factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesener, M.S.; Turley, H.; Allen, W.E.; Willam, C.; Eckardt, K.U.; Talks, K.L.; Wood, S.M.; Gatter, K.C.; Harris, A.L.; Pugh, C.W.; et al. Induction of endothelial PAS domain protein-1 by hypoxia: Characteri-zation and comparison with hypoxia-inducible factor-1alpha. Blood 1998, 92, 2260–2268. [Google Scholar] [CrossRef]
- Semenza, G.; Roth, P.; Fang, H.; Wang, G. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Oktay, Y.; Dioum, E.; Matsuzaki, S.; Ding, K.; Yan, L.-J.; Haller, R.G.; Szweda, L.I.; Garcia, J.A. Hypoxia-inducible Factor 2α Regulates Expression of the Mitochondrial Aconitase Chaperone Protein Frataxin. J. Biol. Chem. 2007, 282, 11750–11756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scortegagna, M.; Ding, K.; Oktay, Y.; Gaur, A.; Thurmond, F.; Yan, L.J.; Marck, B.T.; Matsumoto, A.M.; Shelton, J.M.; Richardson, J.A.; et al. Multiple organ pathology, metabolic abnormalities and impaired home-ostasis of reactive oxygen species in Epas1-/- mice. Nat. Genet. 2003, 35, 331–340. [Google Scholar] [CrossRef]
- Catrina, S.B.; Okamoto, K.; Pereira, T.; Brismar, K.; Poellinger, L. Hyperglycemia regulates hypoxia-inducible fac-tor-1alpha protein stability and function. Diabetes 2004, 53, 3226–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katavetin, P.; Miyata, T.; Inagi, R.; Tanaka, T.; Sassa, R.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible fac-tor/hypoxia-responsible element pathway. J. Am. Soc. Nephrol. 2006, 17, 1405–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunton, J.E.; Kulkarni, R.N.; Yim, S.; Okada, T.; Hawthorne, W.J.; Tseng, Y.H.; Roberson, R.S.; Ricordi, C.; O’Connell, P.J.; Gonzalez, F.J.; et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 2005, 122, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factor 1 (HIF-1) Pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef] [PubMed]
- Willam, C.; Maxwell, P.; Nichols, L.; Lygate, C.; Tian, Y.M.; Bernhardt, W.; Wiesener, M.; Ratcliffe, P.; Eckardt, K.-U.; Pugh, C. HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligation. J. Mol. Cell. Cardiol. 2006, 41, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.-H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Aguila, H.L.; Parikh, N.S.; Li, X.; Lamothe, K.; Duan, L.-J.; Takeda, H.; Lee, F.S.; Fong, G.-H. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 2008, 111, 3229–3235. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Zeng, H.; Roman, R.J.; Chen, J.-X. Inhibition of prolyl hydroxylases alters cell metabolism and reverses pre-existing diastolic dysfunction in mice. Int. J. Cardiol. 2018, 272, 281–287. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-X.; Li, L.; Cantrell, A.C.; Williams, Q.A.; Zeng, H. High Glucose Activates Prolyl Hydroxylases and Disrupts HIF-α Signaling via the P53/TIGAR Pathway in Cardiomyocyte. Cells 2023, 12, 1060. https://doi.org/10.3390/cells12071060
Chen J-X, Li L, Cantrell AC, Williams QA, Zeng H. High Glucose Activates Prolyl Hydroxylases and Disrupts HIF-α Signaling via the P53/TIGAR Pathway in Cardiomyocyte. Cells. 2023; 12(7):1060. https://doi.org/10.3390/cells12071060
Chicago/Turabian StyleChen, Jian-Xiong, Lanfang Li, Aubrey C. Cantrell, Quinesha A. Williams, and Heng Zeng. 2023. "High Glucose Activates Prolyl Hydroxylases and Disrupts HIF-α Signaling via the P53/TIGAR Pathway in Cardiomyocyte" Cells 12, no. 7: 1060. https://doi.org/10.3390/cells12071060
APA StyleChen, J.-X., Li, L., Cantrell, A. C., Williams, Q. A., & Zeng, H. (2023). High Glucose Activates Prolyl Hydroxylases and Disrupts HIF-α Signaling via the P53/TIGAR Pathway in Cardiomyocyte. Cells, 12(7), 1060. https://doi.org/10.3390/cells12071060