

A Knockout of Poly(ADP-Ribose) Polymerase 1 in a Human Cell Line: An Influence on Base Excision Repair Reactions in Cellular Extracts

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cultivation of Cells
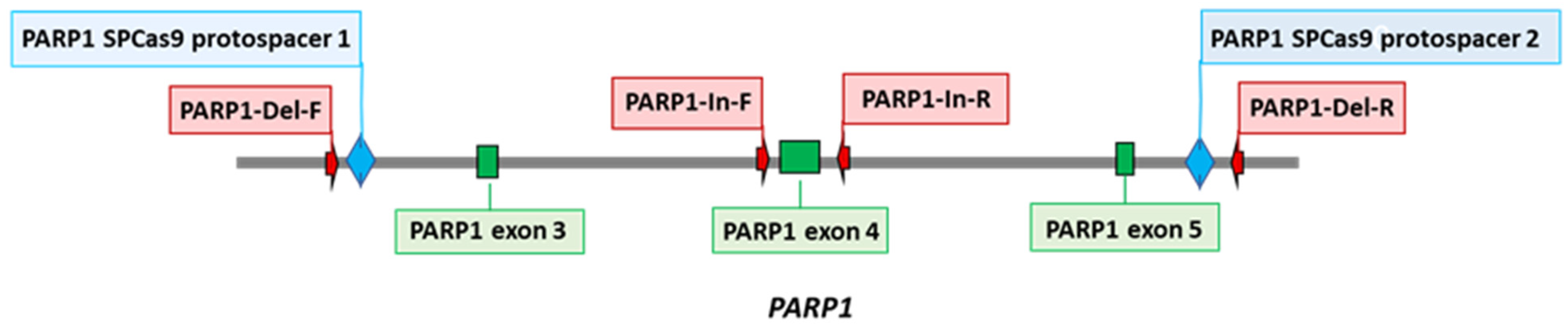
2.3. Creation of PARP1 Knockout Cells (HEK293FT/P1-KO)
2.4. DNA Sequencing
2.5. Estimation of Doubling Time
2.6. Cell Culture Cytotoxicity Assay
2.7. 32P-Labeling of Oligonucleotides and Preparation of BER DNA Substrates
2.8. Isolation of Total RNA
2.9. qPCR Analysis of mRNA Levels in HEK293FT WT and HEK293FT/P1-KO Cells
2.10. WCEs
2.11. Western Blot Analysis of PARP1 in the WCEs of HEK293FT WT and HEK293FT/P1-KO Cells
2.12. Synthesis of [32P]NAD+
2.13. A PARP Activity Assay
2.14. Quantification of the Results of Autoradiography
2.15. Preparation of DNA Substrates Containing AP Sites or 5ʹ-dRP Residues
2.16. Tests for BER Enzymatic Activities in the WCEs
2.16.1. An Assay of Uracil Excision Activity
2.16.2. An Assay of AP Site Cleavage Activity
2.16.3. An Assay of DNA Polymerase Activity
2.16.4. An Assay of DNA Ligase Activity
2.17. A Cell Culture Cytotoxicity Assay
3. Results and Discussion
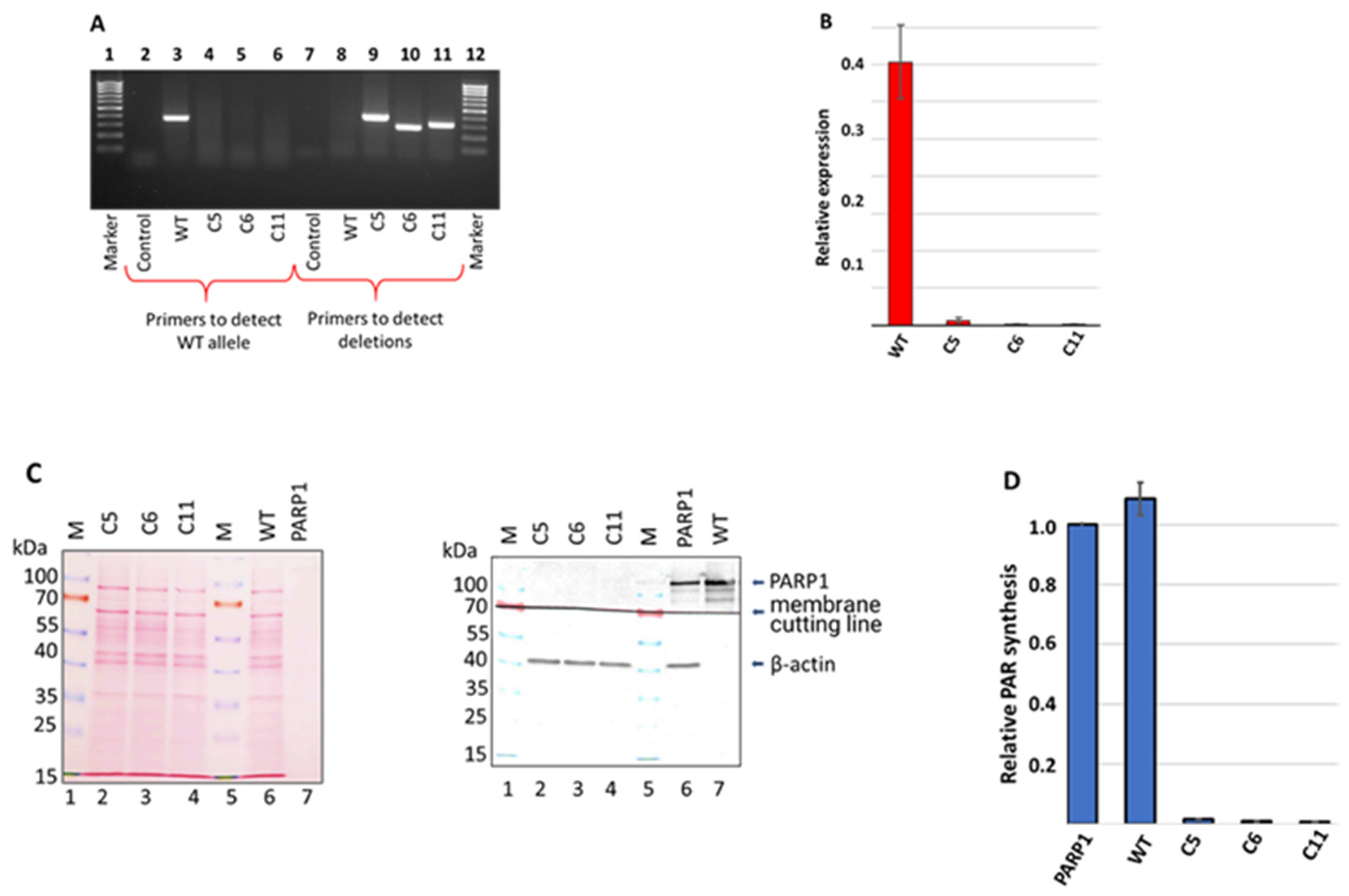
3.1. Generation and Characterization of HEK293FT/P1-KO Cells
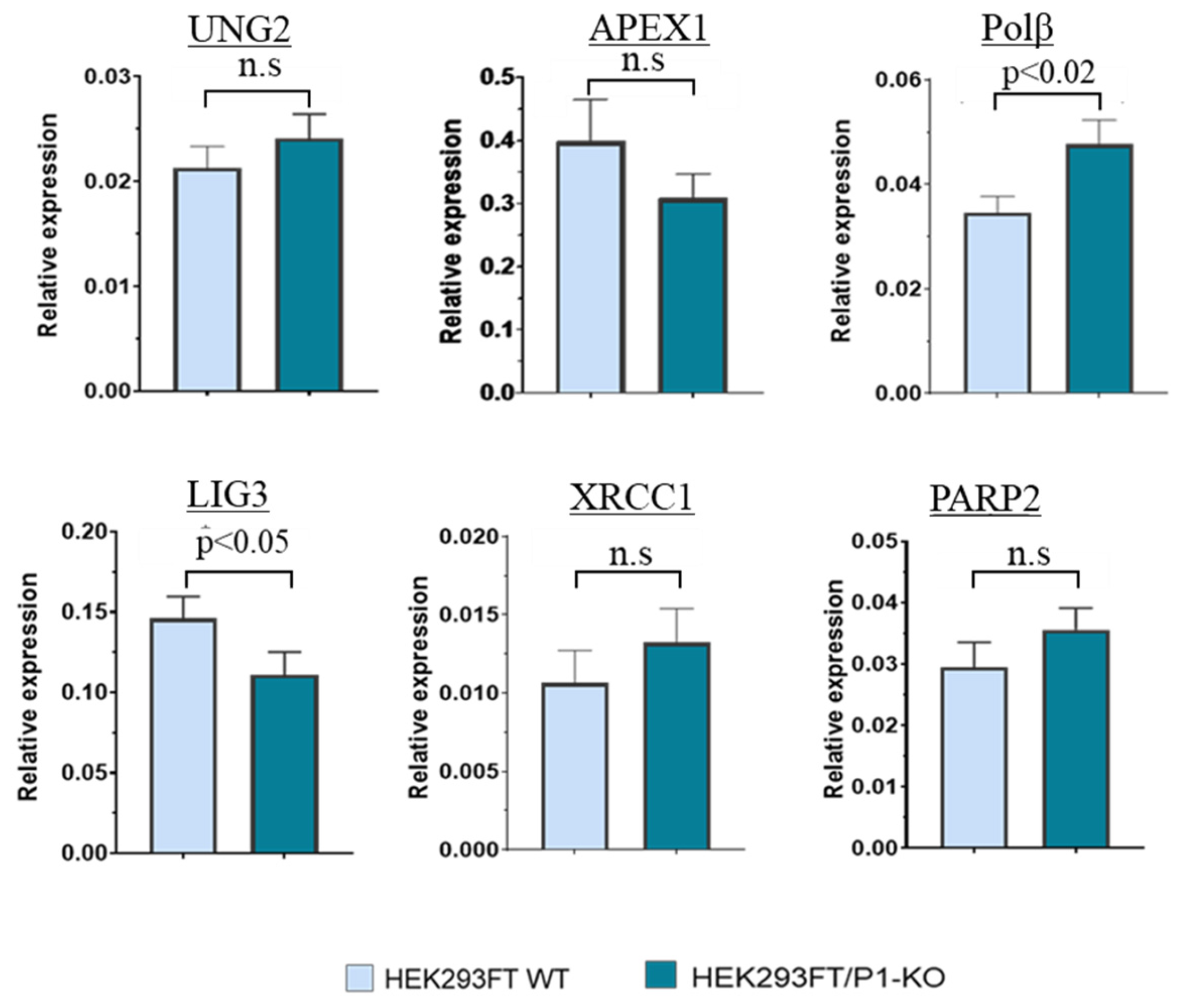
3.2. The Comparison of PARP2, UNG2, APEX1, POLΒ, and XRCC1 mRNA Relative Expression Levels
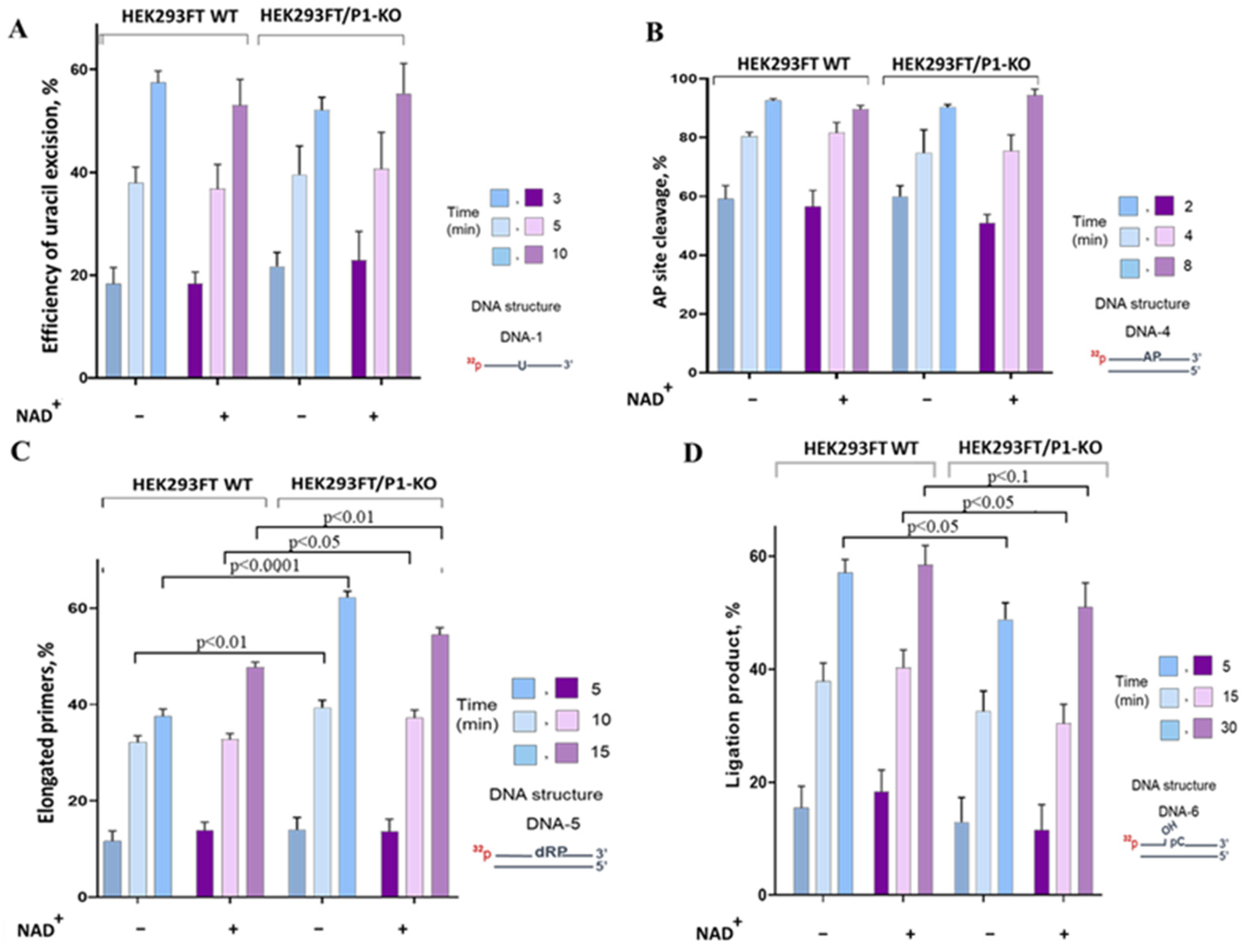
3.3. The Influence of PARP1 Presence in WCEs on the Efficiency of BER Reactions
Uracil Removal
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lüscher, B.; Ahel, I.; Altmeyer, M.; Asworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J. 2021, 289, 7399–7410. [Google Scholar] [CrossRef]
- Lavrik, O.I. PARPs’ impact on base excision DNA repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef]
- Lüscher, B.; Bütepage, M.; Eckei, L.; Krieg, S.; Verheugd, P.; Shilton, B.H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018, 118, 1092–1136. [Google Scholar] [CrossRef]
- Martin-Hernandez, K.; Rodriguez-Vargas, J.M.; Schreiber, V.; Dantzer, F. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Cell Dev. Biol. 2017, 63, 92–101. [Google Scholar] [CrossRef]
- Azarm, K.; Smith, S. Nuclear PARPs and genome integrity. Genes Dev. 2020, 34, 285–301. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, A.; Mir, K.U.I.; Yadav, V.; Chauhan, S.S. Pleiotropic role of PARP1: An overview. 3 Biotech. 2022, 12, 3. [Google Scholar] [CrossRef]
- Ke, Y.; Zhang, J.; Lv, X.; Zeng, X.; Ba, X. Novel insights into PARPs in gene expression: Regulation of RNA metabolism. Cell. Mol. Life Sci. 2019, 76, 3283–3299. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Eleazer, R.; Fondufe-Mittendorf, Y.N. The multifaceted role of PARP1 in RNA biogenesis. Wiley Interdiscip. Rev. RNA 2021, 12, e1617. [Google Scholar] [CrossRef]
- Langelier, M.F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes: Regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef]
- Suskiewicz, M.J.; Palazzo, L.; Hughes, R.; Ahel, I. Progress and outlook in studying the substrate specificities of PARPs and related enzymes. FEBS J. 2021, 288, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, E.A.; Mathbout, L.F.; Fondufe-Mittendorf, Y.N. PARP1 is a versatile factor in the regulation of mRNA stability and decay. Sci. Rep. 2019, 91, 3722. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Ren, E.C. Functional aspects of PARP1 in DNA repair and transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. Chemistry and biology of DNA repair. Angew. Chem. Int. Ed. Engl. 2003, 42, 2946–2974. [Google Scholar] [CrossRef]
- Frosina, G.; Fortini, P.; Rossi, O.; Carrozzino, F.; Raspaglio, G.; Cox, L.S.; Lane, D.P.; Abbondandolo, A.; Dogliotti, E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996, 271, 9573–9578. [Google Scholar] [CrossRef]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar] [CrossRef]
- Gohil, D.; Sarker, A.H.; Roy, R. Base Excision Repair: Mechanisms and Impact in Biology, Disease, and Medicine. Int. J. Mol. Sci. 2023, 24, 14186. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian DNA base excision repair: Dancing in the moonlight. DNA Repair 2020, 93, 102921. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef]
- Baiken, Y.; Kanayeva, D.; Taipakova, S.; Groisman, R.; Ishchenko, A.A.; Begimbetova, D.; Matkarimov, B.; Saparbaev, M. Role of base excision repair pathway in the processing of complex DNA damage generated by oxidative stress and anticancer drugs. Front. Cell Dev. Biol. 2021, 22, 617884. [Google Scholar] [CrossRef]
- Dierich, A.; LeMeur, M.; Walztinger, C.; Chambon, P.; de Murcia, G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [PubMed]
- De Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dantzer, F.; Schreiber, V.; et al. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie 1999, 81, 69–75. [Google Scholar] [CrossRef]
- Shall, S.; de Murcia, G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. 2000, 460, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.J.; Lindahl, T. Down-regulation of DNA repair synthesis at DNA single-strand interruptions in poly(ADP-ribose) polymerase-1 deficient murine cell extracts. DNA Repair 2002, 1, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, M.M.; Belousova, E.A.; Kurgina, T.A.; Ukraintsev, A.A.; Vasil’eva, I.A.; Khodyreva, S.N.; Lavrik, O.I. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci. Rep. 2021, 11, 4849. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.; Khodyreva, S.; Lavrik, O. Poly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase beta in long patch base excision repair. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 685, 80–89. [Google Scholar] [CrossRef]
- Ilina, E.S.; Kochetkova, A.S.; Belousova, E.A.; Kutuzov, M.M.; Lavrik, O.I.; Khodyreva, S.N. Influence of the poly(ADP-ribose) polymerase 1 level on the status of base excision repair in human cells. Mol. Biol. 2023, 57, 285–298. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Malakhova, A.A.; Zakharenko, A.L.; Okorokova, L.S.; Shtokalo, D.N.; Pavlova, S.V.; Medvedev, S.P.; Zakian, S.M.; Nushtaeva, A.A.; Tupikin, A.E.; et al. Transcriptomic analysis of CRISPR/Cas9-mediated PARP1-knockout cells under the influence of topotecan and TDP1 inhibitor. Int. J. Mol. Sci. 2023, 24, 5148. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Malakhova, A.A.; Dyrkheeva, N.S.; Okorokova, L.S.; Medvedev, S.P.; Zakian, S.M.; Kabilov, M.R.; Tupikin, A.A.; Lavrik, O.I. PARP1 gene knockout suppresses expression of DNA base excision repair genes. Dokl. Biochem. Biophys. 2023, 508, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Xu, W.W.; Jiang, S.; Yu, H.; Poon, H.F. The Scattered Twelve Tribes of HEK293. Biomed. Pharmacol. J. 2018, 11, 621–623. [Google Scholar] [CrossRef]
- Kumar, A.; Widen, S.G.; Williams, K.R.; Kedar, P.; Karpel, R.L.; Wilson, S.H. Studies of the domain structure of mammalian DNA polymerase. Identification of a discrete template binding domain. J. Biol. Chem. 1990, 265, 2124–2131. [Google Scholar] [CrossRef]
- Strauss, P.R.; Beard, W.A.; Patterson, T.A.; Wilson, S.H. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 1997, 272, 1302–1307. [Google Scholar] [CrossRef]
- Amé, J.C.; Kalisch, T.; Dantzer, F.; Schreiber, V. Purification of recombinant poly(ADP-ribose) polymerases. Methods Mol. Biol. 2011, 780, 135–152. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. 2010, 6, pdb-prot5439. [Google Scholar] [CrossRef]
- Biade, S.; Sobol, R.W.; Wilson, S.H.; Matsumoto, Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J. Biol. Chem. 1998, 273, 898–902. [Google Scholar] [CrossRef]
- Bradford, M.A. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 277, 680–685. [Google Scholar] [CrossRef]
- Kosova, A.A.; Kutuzov, M.M.; Evdokimov, A.N.; Ilina, E.S.; Belousova, E.A.; Romanenko, S.A.; Trifonov, V.A.; Khodyreva, S.N.; Lavrik, O.I. Poly(ADP-ribosyl)ation and DNA repair synthesis in the extracts of naked mole rat, mouse, and human cells. Aging 2019, 11, 2852–2873. [Google Scholar] [CrossRef]
- Parsons, J.L.; Dianova, I.I.; Allinson, S.L.; Dianov, G.L. Poly(ADP-ribose) polymerase-1 protects excessive DNA strand breaks from deterioration during repair in human cell extracts. FEBS J. 2005, 272, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Williams, J.G.; Hou, E.W.; Wilson, S.H. Pol β associated complex and base excision repair factors in mouse fibroblasts. Nucleic Acids Res. 2012, 40, 11571–11582. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Horton, J.K.; Dai, D.; Wilson, S.H. Repair pathway for PARP-1 DNA-protein crosslinks. DNA Repair 2019, 73, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.V.; Khodyreva, S.N.; Lebedeva, N.A.; Prasad, R.; Wilson, S.H.; Lavrik, O.I. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: Interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005, 33, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, S.; Hartwig, A. PARP1 Is Required for ATM-Mediated p53 Activation and p53-Mediated Gene Expression after Ionizing Radiation. Chem. Res. Toxicol. 2020, 33, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.N.; Chen, H.D.; Tian, C.Q.; Wang, Y.Q.; Miao, Z.H. Polymerase independent repression of FoxO1 transcription by sequence-specific PARP1 binding to FoxO1 promoter. Cell Death Dis. 2020, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.D.; Chen, C.H.; Wang, Y.T.; Guo, N.; Tian, Y.N.; Huan, X.J.; Song, S.S.; He, J.X.; Miao, Z.H. Increased PARP1-DNA binding due to autoPARylation inhibition of PARP1 on DNA rather than PARP1-DNA trapping is correlated with PARP1 inhibitor’s cytotoxicity. Int. J. Cancer. 2019, 145, 714–727. [Google Scholar] [CrossRef]
- Kim, C.; Wang, X.D.; Yu, Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 2020, 26, e60637. [Google Scholar] [CrossRef]
- Aberle, L.; Krüger, A.; Reber, J.M.; Lippmann, M.; Hufnagel, M.; Schmalz, M.; Trussina, I.R.E.A.; Schlesiger, S.; Zubel, T.; Schütz, K.; et al. PARP1 catalytic variants reveal branching and chain length-specific functions of poly(ADP-ribose) in cellular physiology and stress response. Nucleic Acids Res. 2020, 48, 10015–10033. [Google Scholar] [CrossRef]
- Rank, L.; Veith, S.; Gwosch, E.C.; Demgenski, J.; Ganz, M.; Jongmans, M.C.; Vogel, C.; Fischbach, A.; Buerger, S.; Fischer, J.M.; et al. Analyzing structure-function relationships of artificial and cancer-associated PARP1 variants by reconstituting TALEN-generated HeLa PARP1 knock-out cells. Nucleic Acids Res. 2016, 44, 10386–10405. [Google Scholar] [CrossRef]
- Reber, J.M.; Božić-Petković, J.; Lippmann, M.; Mazzardo, M.; Dilger, A.; Warmers, R.; Bürkle, A.; Mangerich, A. PARP1 and XRCC1 exhibit a reciprocal relationship in genotoxic stress response. Cell. Biol. Toxicol. 2023, 39, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.; Andreieva, S.; Korets, K.; Mykytenko, D.; Huleyuk, N.; Vassetzky, Y.; Kavsan, V. Step-wise and punctuated genome evolution drive phenotype changes of tumor cells. Mutat. Res. 2015, 771, 56–69. [Google Scholar] [CrossRef]
- Binz, R.L.; Tian, E.; Sadhukhan, V.; Zhou, D.; Hauer-Jensen, M.; Pathak, R. Identification of novel breakpoints for locus- and region-specific translocations in 293 cells by molecular cytogenetics before and after irradiation. Sci. Rep. 2019, 9, 10554. [Google Scholar] [CrossRef] [PubMed]
- Doseth, B.; Ekre, C.; Slupphaug, G.; Krokan, H.E.; Kavli, B. Strikingly different properties of uracil DNA glycosylases UNG2 and SMUG1 may explain divergent roles in processing of genomic uracil. DNA Repair 2012, 11, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Doseth, B.; Visnes, T.; Wallenius, A.; Ericsson, I.; Sarno, A.; Pettersen, H.S.; Flatberg, A.; Catterall, T.; Slupphaug, G.; Krokan, H.E.; et al. Uracil-DNA glycosylase in base excision repair and adaptive immunity: Species differences between man and mouse. J. Biol. Chem. 2011, 286, 16669–16680. [Google Scholar] [CrossRef] [PubMed]
- Khodyreva, S.; Lavrik, O. Non-canonical interaction of DNA repair proteins with intact and cleaved AP sites. DNA Repair 2020, 90, 102847. [Google Scholar] [CrossRef]
- Mol, C.D.; Hosfield, D.J.; Tainer, J.A. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: The 3′ ends justify the means. Mutat. Res. 2000, 460, 211–229. [Google Scholar] [CrossRef]
- Wilson, D.M.; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Caldecott, K.W. XRCC1 protein; form and function. DNA Repair 2019, 81, 102664. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Endutkin, A.V.; Yudkina, A.V.; Sidorenko, V.S.; Zharkov, D.O.J. Transient protein-protein complexes in base excision repair. Biomol. Struct. Dyn. 2019, 37, 4407–4418. [Google Scholar] [CrossRef] [PubMed]
- Kurgina, T.A.; Moor, N.A.; Kutuzov, M.M.; Lavrik, O.I. The HPF1-dependent histone PARylation catalyzed by PARP2 is specifically stimulated by an incised AP site-containing BER DNA intermediate. DNA Repair 2022, 120, 103423. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.D.; Xu, J.; Gunasekera, K.; Schürmann, D.; Vågbø, C.B.; Ferrari, E.; Slupphaug, G.; Hottiger, M.O.; Schär, P.; Steinacher, R. Covalent PARylation of DNA base excision repair proteins regulates DNA demethylation. Nat. Commun. 2024, 15, 184. [Google Scholar] [CrossRef]
- Parsons, J.L.; Dianov, G.L. Co-ordination of base excision repair and genome stability. DNA Repair 2013, 12, 326–333. [Google Scholar] [CrossRef]
- Edmonds, M.J.; Parsons, J.L. Regulation of base excision repair proteins by ubiquitylation. Exp. Cell. Res. 2014, 329, 132–138. [Google Scholar] [CrossRef]
- Carter, R.J.; Parsons, J.L. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell. Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Structure |
|---|---|---|
| DNA-1 | 5′-pGGGAGGCCCTGGCGTTUCCCGGCTTAGTCGCC-3′ |  |
| DNA-2 | 5′-pGGCGACTAAGCCGGG-3′ |  |
| DNA-3 | 5′-pGGGAGGCCCTGGCGTTUCCCGGCTTAGTCGCC 3′-CCCTCCGGGACCGCAAGGGGCCGAATCAGCGG |  |
| DNA-4 | 5′-pGGCGACTAAGCCGGG pUCCCGGCTTAGTCGCC 3′-CCGCTGATTCGGCCCGTTGCGGTCCCGGGCGG |  |
| DNA-5 | 5′-pGGCGACTAAGCCGGG pCAACGCCAGGGCCTCCC 3′-CCGCTGATTCGGCCCGTTGCGGTCCCGGAGGG |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodyreva, S.N.; Ilina, E.S.; Dyrkheeva, N.S.; Kochetkova, A.S.; Yamskikh, A.A.; Maltseva, E.A.; Malakhova, A.A.; Medvedev, S.P.; Zakian, S.M.; Lavrik, O.I. A Knockout of Poly(ADP-Ribose) Polymerase 1 in a Human Cell Line: An Influence on Base Excision Repair Reactions in Cellular Extracts. Cells 2024, 13, 302. https://doi.org/10.3390/cells13040302
Khodyreva SN, Ilina ES, Dyrkheeva NS, Kochetkova AS, Yamskikh AA, Maltseva EA, Malakhova AA, Medvedev SP, Zakian SM, Lavrik OI. A Knockout of Poly(ADP-Ribose) Polymerase 1 in a Human Cell Line: An Influence on Base Excision Repair Reactions in Cellular Extracts. Cells. 2024; 13(4):302. https://doi.org/10.3390/cells13040302
Chicago/Turabian StyleKhodyreva, Svetlana N., Ekaterina S. Ilina, Nadezhda S. Dyrkheeva, Alina S. Kochetkova, Alexandra A. Yamskikh, Ekaterina A. Maltseva, Anastasia A. Malakhova, Sergey P. Medvedev, Suren M. Zakian, and Olga I. Lavrik. 2024. "A Knockout of Poly(ADP-Ribose) Polymerase 1 in a Human Cell Line: An Influence on Base Excision Repair Reactions in Cellular Extracts" Cells 13, no. 4: 302. https://doi.org/10.3390/cells13040302
APA StyleKhodyreva, S. N., Ilina, E. S., Dyrkheeva, N. S., Kochetkova, A. S., Yamskikh, A. A., Maltseva, E. A., Malakhova, A. A., Medvedev, S. P., Zakian, S. M., & Lavrik, O. I. (2024). A Knockout of Poly(ADP-Ribose) Polymerase 1 in a Human Cell Line: An Influence on Base Excision Repair Reactions in Cellular Extracts. Cells, 13(4), 302. https://doi.org/10.3390/cells13040302