

Foot-and-Mouth Disease Virus Capsid Protein VP1 Antagonizes Type I Interferon Signaling via Degradation of Histone Deacetylase 5

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Infection
2.2. Reagents, Antibodies, and Plasmids
2.3. Coimmunoprecipitation and Western Blotting
2.4. Reporter Gene Assays
2.5. RNA Extraction and RT-qPCR
2.6. Cell Treatments, Transfection, and ELISA
2.7. RNA Interference
2.8. Statistical Analysis
3. Results
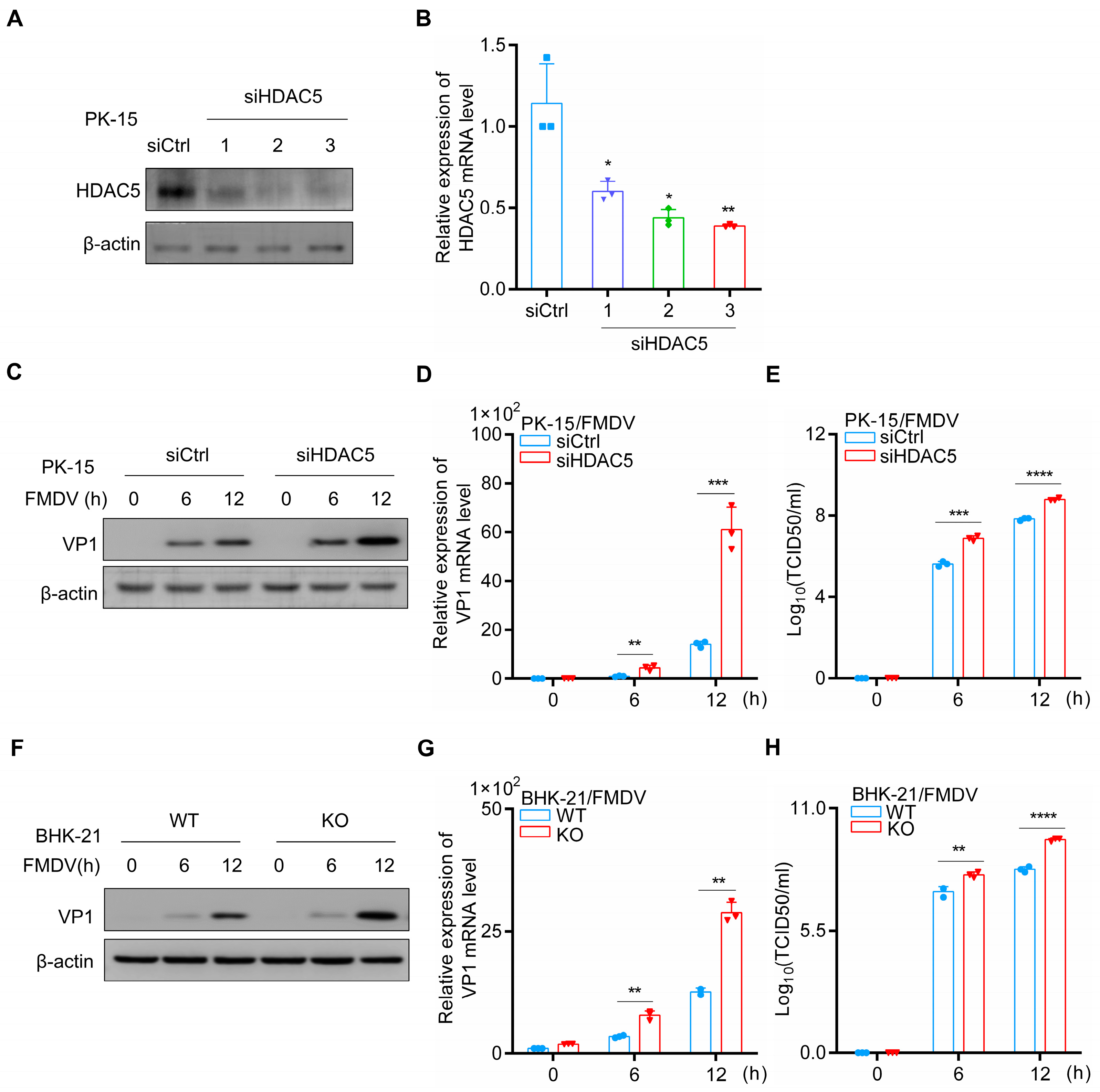
3.1. HDAC5 Inhibits FMDV Replication
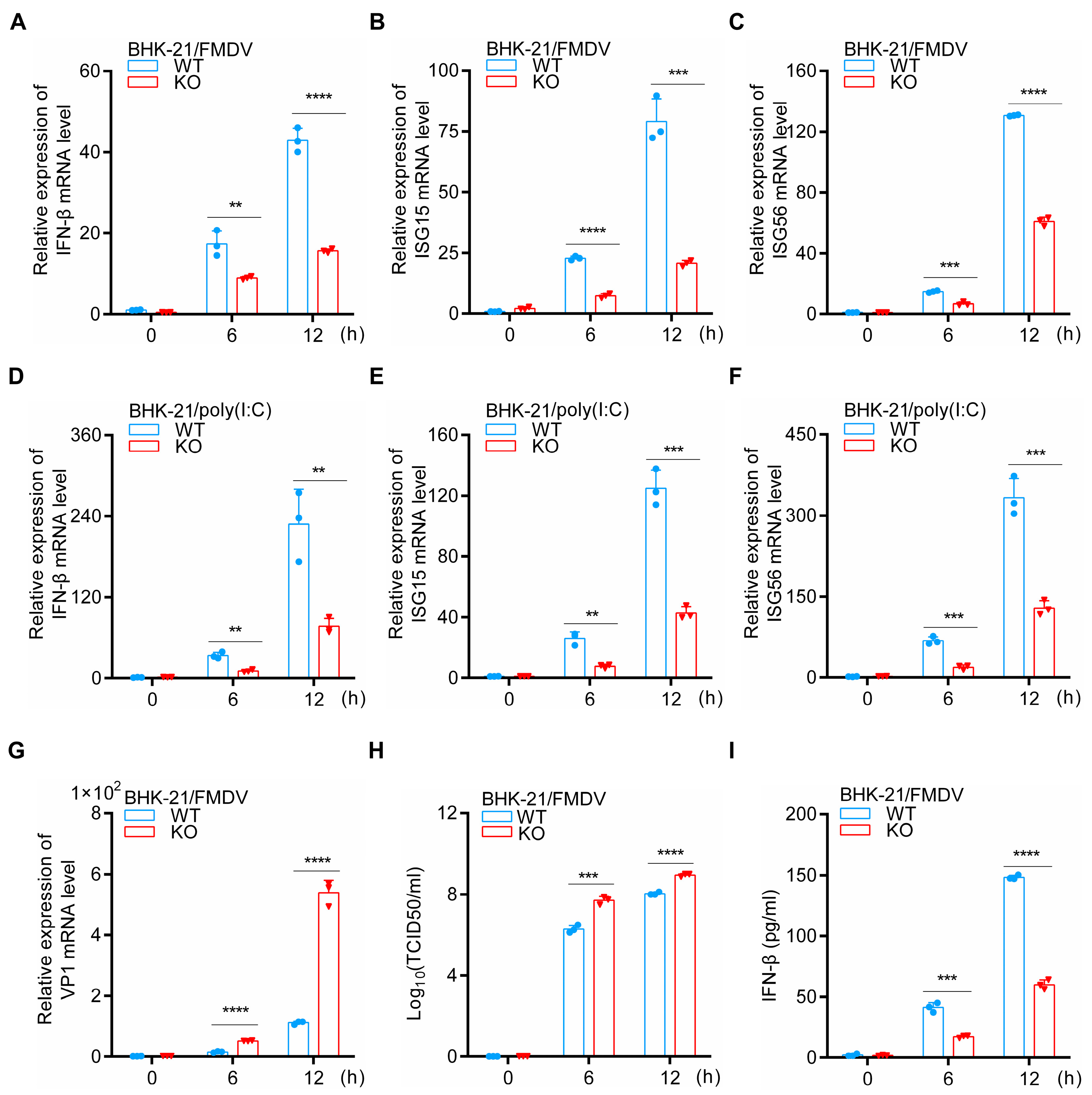
3.2. HDAC5 Enhances IFN-β and ISGs Expression during FMDV Infection
3.3. HDAC5 Regulates FMDV-Induced Phosphorylation of IRF3
3.4. FMDV Capsid Protein VP1 Targets and Degrades HDAC5 through the Proteasome Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Knowles, N.J.; Samuel, A.R. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 2003, 91, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.; Belsham, G.J. Foot-and-mouth disease: Past, present and future. Vet. Res. 2013, 44, 116. [Google Scholar] [CrossRef]
- Knight-Jones, T.J.D.; Robinson, L.; Charleston, B.; Rodriguez, L.L.; Gay, C.G.; Sumption, K.J.; Vosloo, W. Global Foot-and-Mouth Disease Research Update and Gap Analysis: 2-Epidemiology, Wildlife and Economics. Transbound. Emerg. Dis. 2016, 63 (Suppl. 1), 14–29. [Google Scholar] [CrossRef] [PubMed]
- Casey-Bryars, M.; Reeve, R.; Bastola, U.; Knowles, N.J.; Auty, H.; Bachanek-Bankowska, K.; Fowler, V.L.; Fyumagwa, R.; Kazwala, R.; Kibona, T.; et al. Waves of endemic foot-and-mouth disease in eastern Africa suggest feasibility of proactive vaccination approaches. Nat. Ecol. Evol. 2018, 2, 1449–1457. [Google Scholar] [CrossRef]
- König, G.A.; Palma, E.L.; Maradei, E.; Piccone, M.E. Molecular epidemiology of foot-and-mouth disease virus types A and O isolated in Argentina during the 2000–2002 epizootic. Vet. Microbiol. 2007, 124, 1–15. [Google Scholar] [CrossRef]
- Klein, J. Understanding the molecular epidemiology of foot-and-mouth-disease virus. Infect. Genet Evol. 2009, 9, 153–161. [Google Scholar] [CrossRef]
- Paton, D.J.; Reeve, R.; Capozzo, A.V.; Ludi, A. Estimating the protection afforded by foot-and-mouth disease vaccines in the laboratory. Vaccine 2019, 37, 5515–5524. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J. Translation and replication of FMDV RNA. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 288, pp. 43–70. [Google Scholar] [CrossRef]
- Carrillo, C.; Tulman, E.R.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.F.; Rock, D.L. Comparative genomics of foot-and-mouth disease virus. J. Virol. 2005, 79, 6487–6504. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, S.Q.; Guo, H.C. Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 2016, 13, 107. [Google Scholar] [CrossRef]
- Li, X.Y.; Wang, J.C.; Liu, J.; Li, Z.H.; Wang, Y.Q.; Xue, Y.F.; Li, X.Q.; Cao, H.; Zheng, S.J.J. Engagement of soluble resistance-related calcium binding protein (sorcin) with foot-and-mouth disease virus (FMDV) VP1 inhibits type I interferon response in cells. Vet. Microbiol. 2013, 166, 35–46. [Google Scholar] [CrossRef]
- Hao, J.H.; Shen, C.C.; Wei, N.N.; Yan, M.H.; Zhang, X.G.; Xu, G.W.; Zhang, D.J.; Hou, J.; Cao, W.J.; Jin, Y.; et al. Foot-and-Mouth Disease Virus Capsid Protein VP1 Antagonizes TPL2-Mediated Activation of the IRF3/IFN-β Signaling Pathway to Facilitate the Virus Replication. Front Immunol. 2021, 12, 686494, Correction to Front Immunol. 2021, 11, 580334. [Google Scholar] [CrossRef]
- Ekanayaka, P.; Lee, S.Y.; Herath, T.U.B.; Kim, J.H.; Kim, T.H.; Lee, H.; Chathuranga, K.; Chathuranga, W.A.G.; Park, J.H.; Lee, J.S. Foot-and-mouth disease virus VP1 target the MAVS to inhibit type-I interferon signaling and VP1 E83K mutation results in virus attenuation. PLoS Pathog. 2020, 16, e1009057. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, F.; Zhu, Z.X.; Yang, Y.; Wang, Z.F.; Cao, W.J.; Dang, W.; Li, L.L.; Mao, R.Q.; Liu, Y.J.; et al. Cellular DNAJA3, a Novel VP1-Interacting Protein, Inhibits Foot-and-Mouth Disease Virus Replication by Inducing Lysosomal Degradation of VP1 and Attenuating Its Antagonistic Role in the Beta Interferon Signaling Pathway. J. Virol. 2019, 93, 10.1128. [Google Scholar] [CrossRef]
- Fan, W.C.; Zhang, D.; Qian, P.; Qian, S.H.; Wu, M.G.; Chen, H.C.; Li, X.M. Swine TRIM21 restricts FMDV infection via an intracellular neutralization mechanism. Antivir. Res. 2016, 127, 32–40. [Google Scholar] [CrossRef]
- Fu, S.Z.; Yang, W.P.; Ru, Y.; Zhang, K.S.; Wang, Y.; Liu, X.T.; Li, D.; Zheng, H.X. DDX56 cooperates with FMDV 3A to enhance FMDV replication by inhibiting the phosphorylation of IRF3. Cell. Signal. 2019, 64, 109393. [Google Scholar] [CrossRef] [PubMed]
- Weerawardhana, A.; Uddin, M.B.; Choi, J.H.; Pathinayake, P.; Shin, S.H.; Chathuranga, K.; Park, J.H.; Lee, J.S. Foot-and-mouth disease virus non-structural protein 2B downregulates the RLR signaling pathway degradation of RIG-I and MDA5. Front Immunol. 2022, 13, 1020262. [Google Scholar] [CrossRef]
- Wu, X.J.; Hu, Y.; Sui, C.; Pan, L.; Yoo, D.W.; Miller, L.C.; Lee, C.; Cong, X.Y.; Li, J.T.; Du, Y.J.; et al. Multiple-Site SUMOylation of FMDV 3C Protease and Its Negative Role in Viral Replication. J. Virol. 2022, 96, e0061222. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Abdullah, S.W.; Wu, J.E.; Tang, J.L.; Zhang, Y.; Dong, H.; Bai, M.Y.; Wei, S.M.; Sun, S.Q.; Guo, H.C. Foot-and-mouth disease virus downregulates vacuolar protein sorting 28 to promote viral replication. J. Virol. 2023, 97, e00181-23. [Google Scholar] [CrossRef] [PubMed]
- Guise, A.J.; Budayeva, H.G.; Diner, B.A.; Cristea, I.M. Histone Deacetylases in Herpesvirus Replication and Virus-Stimulated Host Defense. Viruses 2013, 5, 1607–1632. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.F.; Lupiani, B.; AI-Mahmood, M.; Reddy, S.M. Marek’s disease virus U3 protein kinase phosphorylates chicken HDAC 1 and 2 and regulates viral replication and pathogenesis. PLoS Pathog. 2021, 17, e1009307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Han, W.J.; Li, M.Z.; Bai, R.M.; Tian, Z.Y.; Yuan, W.Z.; Li, L.M. Histone acetylation regulates BMMCs recognition of foot-and-mouth disease virus-like particles. Int. Immunopharmacol. 2023, 121, 2869–2883. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.T.; Chen, H.T.; Liu, X.S.; Wang, Y.X.; Fan, A.X.; Qi, L.L.; Pan, L.; Bai, W.L.; Zhang, Y.G.; Sun, Y.F. ID1 inhibits foot-and-mouth disease virus replication via targeting of interferon pathways. FEBS J. 2021, 288, 4364–4381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Wang, X.W.; Qu, M.; Li, Z.Y.; Yin, X.P.; Tang, L.J.; Liu, X.T.; Sun, Y.F. Foot-and-mouth disease virus structural protein VP3 interacts with HDAC8 and promotes its autophagic degradation to facilitate viral replication. Autophagy 2023, 19, 2869–2883. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, X.G.; Zhang, P.; Li, D.; Xu, S.; Zhou, Q.Q.; Guo, M.; Huai, W.W.; Chen, X.; Wang, Q.X.; et al. Rb selectively inhibits innate IFN-beta production by enhancing deacetylation of IFN-beta promoter through HDACI and HDAC8. J. Autoimmun. 2016, 73, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Stuart, J.H.; Talbot-Cooper, C.; Agrawal-Singh, S.; Huntly, B.; Smid, A.I.; Snowden, J.S.; Dupont, L.; Smith, G.L. Histone deacetylase 4 promotes type I interferon signaling, restricts DNA viruses; is degraded via vaccinia virus protein C6. Proc. Natl. Acad. Sci. USA 2019, 116, 11997–12006. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.C.; Lei, C.Q.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef]
- Sen, A.; Pruijssers, A.J.; Dermody, T.S.; García-Sastre, A.; Greenberg, H.B. The Early Interferon Response to Rotavirus Is Regulated by PKR and Depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 2011, 85, 3717–3732. [Google Scholar] [CrossRef]
- Chen, D.D.; Jiang, J.Y.; Lu, L.F.; Zhang, C.; Zhou, X.Y.; Li, Z.C.; Zhou, Y.; Li, S. Zebrafish Uba1 Degrades IRF3 through K48-Linked Ubiquitination to Inhibit IFN Production. J. Immunol. 2021, 207, 512–522. [Google Scholar] [CrossRef]
- De los Santos, T.; Botton, S.D.; Weiblen, R.; Grubman, M.J. The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J. Virol. 2006, 80, 1906–1914. [Google Scholar] [CrossRef]
- Li, D.; Yang, W.P.; Yang, F.; Liu, H.A.; Zhu, Z.X.; Lian, K.Q.; Lei, C.Q.; Li, S.; Liu, X.T.; Zheng, H.X.; et al. The VP3 structural protein of foot-and-mouth disease virus inhibits the IFN-β signaling pathway. FASEB J. 2016, 30, 1757–1766. [Google Scholar] [CrossRef]
- Lomonte, P.; Thomas, J.; Texier, P.; Caron, C.; Khochbin, S.; Epstein, A.L. Functional interaction between class II histone deacetylases and ICPO of herpes simplex virus type 1. J. Virol. 2004, 78, 6744–6757. [Google Scholar] [CrossRef]
- Galvin, H.D.; Husain, M. Influenza A virus-induced host caspase and viral PA-X antagonize the antiviral host factor, histone deacetylase 4. J. Biol. Chem. 2019, 294, 20207–20221. [Google Scholar] [CrossRef]
- Hou, S.T.; Wang, X.W.; Ren, S.H.; Meng, X.L.; Yin, X.P.; Zhang, J.; Tarasiuk, K.; Pejsak, Z.; Jiang, T.; Mao, R.Q.; et al. Knockout of HDAC9 Gene Enhances Foot-and-Mouth Disease Virus Replication. Front. Microbiol. 2022, 13, 805606. [Google Scholar] [CrossRef]
- Moon, J.; Kaowinn, S.; Cho, I.R.; Min, D.S.; Myung, H.; Oh, S.; Kaewpiboon, C.; Kraemer, O.H.; Chung, Y.H. Hepatitis C virus core protein enhances hepatocellular carcinoma cells to be susceptible to oncolytic vesicular stomatitis virus through down-regulation of HDAC4. Biochem. Bioph. Res. Commun. 2016, 474, 428–434. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Ding, Y.Y.; Liu, Y.Q.; Zhao, D.Z.; Zhao, K.; Shen, Q.C.; Liu, X.G.; Zhu, X.H.; Li, N.; et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol. 2016, 17, 1005, Correction to Nat. Immunol. 2016, 17, 806. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Li, F.; Choi, U.Y.; Yu, H.R.; Aldrovandi, G.M.; Feng, P.H.; Gao, S.J.; Hong, Y.K.; Jung, J.U. Deregulation of HDAC5 by Viral Interferon Regulatory Factor 3 Plays an Essential Role in Kaposi’s Sarcoma-Associated Herpesvirus-Induced Lymphangiogenesis. mBio 2018, 9, e02217-17. [Google Scholar] [CrossRef] [PubMed]
- Soday, L.; Lu, Y.X.; Albarnaz, J.D.; Davies, C.T.R.; Antrobus, R.; Smith, G.L.; Weekes, M.P. Quantitative Temporal Proteomic Analysis of Vaccinia Virus Infection Reveals Regulation of Histone Deacetylases by an Interferon Antagonist. Cell Rep. 2019, 27, 1920–1933.e7. [Google Scholar] [CrossRef]
- Taha, T.Y.; Anirudhan, V.; Limothai, U.; Loeb, D.D.; Petukhov, P.A.; McLachlan, A. Modulation of hepatitis B virus pregenomic RNA stability and splicing by histone deacetylase 5 enhances viral biosynthesis. PLoS Pathog. 2020, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Alexandersen, S.; Zhang, Z.; Donaldson, A.I.; Garland, A.J.M. The pathogenesis and diagnosis of foot-and-mouth disease. J Comp. Pathol. 2003, 129, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, M.E.; El-Deeb, A.H.; Zeidan, S.M.; Hussein, H.A.; Abu-El-Naga, H.I. Enhanced protection against FMDV in cattle after prime- boost vaccination based on mucosal and inactivated FMD vaccine. Vet. Microbiol. 2017, 210, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.Y.; Chen, L.B.; Dong, H.; Li, S.; Zhang, Y.; Yin, S.H.; Tian, Y.F.; Ding, Y.Z.; Sun, S.Q.; Shang, S.B.; et al. Enhanced antigen-specific CD8 T cells contribute to early protection against FMDV through swine DC vaccination. J. Virol. 2024, 98, e0200223. [Google Scholar] [CrossRef] [PubMed]
- Telling, R.C.; Elsworth, R. Submerged culture of hamster kidney cells in a stainless steel vessel. Biotechnol. Bioeng. 1965, 7, 417–434. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence (5′-3′) |
|---|---|
| Human HDAC5 | CCACGCTAGATGAGATCCAGACAG |
| CACAAGGCAGCACAGCATACATC | |
| Human IFN-β | GGACGCCGGACGCCGCATTGACCATCTATG |
| ACAATAGTCTCATTCCAGCCAGTGC | |
| Human GAPDH | GTGACGTTGACATCCGTAAAGA |
| GCCGGACTCATCGTACTCC | |
| Human ISG56 | TCATCAGGTCAAGGATAGTC |
| CCACACTGTATTTGGTGTCTAGG | |
| Human ISG15 | GGAATAACAAGGGCCGCAGCAG |
| AGGTCAGCCAGAACAGGTCGTC | |
| VSV-G | CAAGTCAAAATGCCCAAGAGTCACA |
| TTTCCTTGCATTGTTCTACAGATGG | |
| Mouse IFN-β | CAGCTCCAAGAAAGGACGAAC |
| GGCAGTGTAACTCTTCTGCAT | |
| Mouse ISG15 | TCCTGGTGTCCGTGACTAACTC |
| AAGACCGTCCTGGAGCACTG | |
| Mouse ISG56 | TGAGATGGACTGTGAGGAAGGC |
| TCTTGGCGATAGGCTACGACTG | |
| Mouse GAPDH | AGGTCGGTGTGAACGGATTTG |
| TGTAGACCATGTAGTTGAGGTCA | |
| FMDV-VP1 | GACAACACCACCAACCCA |
| CCTTCTGAGCCAGCACTT | |
| Sus IFN-β | ACCTACAGGGCGGACTTCAA |
| GTCTCATTCCACCCAGTGCT | |
| Sus ISG15 | ATGGGCTGGGACCTGACGG |
| TTAGCTCCGCCCGCCAGGCT | |
| Sus ISG56 | ACGGCTGCCTAATTTACAGC |
| AGTGGCTGATATCTGGGTGC | |
| Sus β-actin | GCTGGCCGGGACCTGACAGACTACC |
| TCTCCAGGGAGGAAGAGGATGCGGC |
| siRNA Name | siRNA Sequence (5′-3′) |
|---|---|
| siHDAC5-1 | GUCCAGUGCUGGUUACAAATT |
| UUUGUAACCAGCACUGGACTT | |
| siHDAC5-2 | GGCAAGUUCAUGAGCACAUTT |
| AUGUGCUCAUGAACUUGCCTT | |
| siHDAC5-3 | CCACGCUAGAGAAAGUCAUTT |
| AUGACUUUCUCUAGCGUGGTT | |
| siRNA control | UUCUCCGAACGUGUCACGUTT |
| ACGUGACACGUUCGGAGAATT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Q.; Ren, S.; Dou, Y.; Tadele, B.A.; Hu, T.; Zhou, L.; Wang, T.; Yao, K.; Xu, J.; Yin, X.; et al. Foot-and-Mouth Disease Virus Capsid Protein VP1 Antagonizes Type I Interferon Signaling via Degradation of Histone Deacetylase 5. Cells 2024, 13, 539. https://doi.org/10.3390/cells13060539
Gong Q, Ren S, Dou Y, Tadele BA, Hu T, Zhou L, Wang T, Yao K, Xu J, Yin X, et al. Foot-and-Mouth Disease Virus Capsid Protein VP1 Antagonizes Type I Interferon Signaling via Degradation of Histone Deacetylase 5. Cells. 2024; 13(6):539. https://doi.org/10.3390/cells13060539
Chicago/Turabian StyleGong, Qing, Shanhui Ren, Yongxi Dou, Berihun Afera Tadele, Tao Hu, Luoyi Zhou, Tao Wang, Kaishen Yao, Jian Xu, Xiangping Yin, and et al. 2024. "Foot-and-Mouth Disease Virus Capsid Protein VP1 Antagonizes Type I Interferon Signaling via Degradation of Histone Deacetylase 5" Cells 13, no. 6: 539. https://doi.org/10.3390/cells13060539
APA StyleGong, Q., Ren, S., Dou, Y., Tadele, B. A., Hu, T., Zhou, L., Wang, T., Yao, K., Xu, J., Yin, X., & Sun, Y. (2024). Foot-and-Mouth Disease Virus Capsid Protein VP1 Antagonizes Type I Interferon Signaling via Degradation of Histone Deacetylase 5. Cells, 13(6), 539. https://doi.org/10.3390/cells13060539