
Human Glial Cells as Innovative Targets for the Therapy of Central Nervous System Pathologies



Abstract
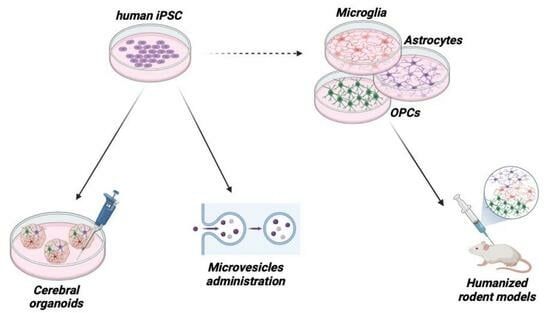
:
1. Heterogeneity and Roles of Glial Cells in Neurodegenerative Pathologies of the Central Nervous System
2. Involvement of Glial Cells in Neurodegenerative Processes: Data from Post-Mortem Human Tissues
3. A Window to Human Glial Cells Reactivity: In Vivo Monitoring of Neuroinflammation
4. Development of Innovative Methods to Study Functional Human Glial Cells
4.1. Human iPSCs
4.2. Cerebral Organoids
4.3. Humanized Mouse Models
5. Glial Cells as Drugs Themselves: Administration of Glia-Derived Microvesicles
6. Drugs Targeting Glial Cells
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia States and Nomenclature: A Field at Its Crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte Contribution to Dysfunction, Risk and Progression in Neurodegenerative Disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Rupareliya, V.P.; Singh, A.A.; Butt, A.M.; A, H.; Kumar, H. The “Molecular Soldiers” of the CNS: Astrocytes, a Comprehensive Review on Their Roles and Molecular Signatures. Eur. J. Pharmacol. 2023, 959, 176048. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, Y.; Leng, S.; Ma, Y.; Jiang, Q.; Wen, Q.; Ju, S.; Hu, J. The Relationship between Inflammation, Impaired Glymphatic System, and Neurodegenerative Disorders: A Vicious Cycle. Neurobiol. Dis. 2024, 192, 106426. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, A.; Ji, R.R. The Similar and Distinct Roles of Satellite Glial Cells and Spinal Astrocytes in Neuropathic Pain. Cells 2023, 12, 965. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, D.; Murashova, L.; Burzak, N.; Dyachuk, V. Satellite Glial Cells: Morphology, Functional Heterogeneity, and Role in Pain. Front. Cell Neurosci. 2022, 16, 10195449. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-Mediated Neuroinflammation in Neurodegenerative Diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Tejera, D.; Heneka, M.T. Microglia in Neurodegenerative Disorders. Methods Mol. Biol. 2019, 2034, 57–67. [Google Scholar]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and Astrocyte Dysfunction in Parkinson’s Disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Glial Cells in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2014, 262 Pt B, 111–120. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Butt, A.; Li, B.; Illes, P.; Zorec, R.; Semyanov, A.; Tang, Y.; Sofroniew, M.V. Astrocytes in Human Central Nervous System Diseases: A Frontier for New Therapies. Signal Transduct. Target. Ther. 2023, 8, 396. [Google Scholar]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cree, B.A.C. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef]
- Sever, B.; Ciftci, H.; Demirci, H.; Sever, H.; Ocak, F.; Yulug, B.; Tateishi, H.; Tateishi, T.; Otsuka, M.; Fujita, M.; et al. Comprehensive Research on Past and Future Therapeutic Strategies Devoted to Treatment of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 2400. [Google Scholar] [CrossRef]
- Colombo, E.; Cordiglieri, C.; Melli, G.; Newcombe, J.; Krumbholz, M.; Parada, L.F.; Medico, E.; Hohlfeld, R.; Meinl, E.; Farina, C. Stimulation of the Neurotrophin Receptor TrkB on Astrocytes Drives Nitric Oxide Production and Neurodegeneration. J. Exp. Med. 2012, 209, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Cappoli, N.; Tabolacci, E.; Aceto, P.; Dello Russo, C. The Emerging Role of the BDNF-TrkB Signaling Pathway in the Modulation of Pain Perception. J. Neuroimmunol. 2020, 349, 577406. [Google Scholar] [CrossRef] [PubMed]
- Mc Guire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear Factor Kappa B (NF-ΚB) in Multiple Sclerosis Pathology. Trends Mol. Med. 2013, 19, 604–613. [Google Scholar] [CrossRef]
- Singh, S.; Singh, T.G. Role of Nuclear Factor Kappa B (NF-ΚB) Signalling in Neurodegenerative Diseases: An Mechanistic Approach. Curr. Neuropharmacol. 2020, 18, 918–935. [Google Scholar] [CrossRef]
- Popiolek-Barczyk, K.; Mika, J. Targeting the Microglial Signaling Pathways: New Insights in the Modulation of Neuropathic Pain. Curr. Med. Chem. 2016, 23, 2908–2928. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.S.; Amici, M.; Bortolotto, Z.A.; Doherty, A.; Csaba, Z.; Fafouri, A.; Dournaud, P.; Gressens, P.; Collingridge, G.L.; Peineau, S. The Role of JAK-STAT Signaling within the CNS. JAKSTAT 2013, 2, e22925. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, C.; Chen, Z.; Wang, X.; Hong, F.; Hao, D. Regulation of the JAK/STAT Signaling Pathway in Spinal Cord Injury: An Updated Review. Front. Immunol. 2023, 14, 1276445. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 Is a Critical Regulator of Astrogliosis and Scar Formation after Spinal Cord Injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. An Introduction to the Roles of Purinergic Signalling in Neurodegeneration, Neuroprotection and Neuroregeneration. Neuropharmacology 2016, 104, 4–17. [Google Scholar] [CrossRef]
- Magni, G.; Riccio, D.; Ceruti, S. Tackling Chronic Pain and Inflammation through the Purinergic System. Curr. Med. Chem. 2018, 25, 3830–3865. [Google Scholar] [CrossRef]
- Sood, A.; Preeti, K.; Fernandes, V.; Khatri, D.K.; Singh, S.B. Glia: A Major Player in Glutamate-GABA Dysregulation-Mediated Neurodegeneration. J. Neurosci. Res. 2021, 99, 3148–3189. [Google Scholar] [CrossRef]
- Matsuka, Y.; Afroz, S.; Dalanon, J.C.; Iwasa, T.; Waskitho, A.; Oshima, M. The Role of Chemical Transmitters in Neuron-Glia Interaction and Pain in Sensory Ganglion. Neurosci. Biobehav. Rev. 2020, 108, 393–399. [Google Scholar] [CrossRef]
- Volonté, C.; Amadio, S.; Fabbrizio, P.; Apolloni, S. Functional Microglia Neurotransmitters in Amyotrophic Lateral Sclerosis. Semin. Cell Dev. Biol. 2019, 94, 121–128. [Google Scholar] [CrossRef]
- Subbarayan, M.S.; Joly-Amado, A.; Bickford, P.C.; Nash, K.R. CX3CL1/CX3CR1 Signaling Targets for the Treatment of Neurodegenerative Diseases. Pharmacol. Ther. 2022, 231, 107989. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chu, J.M.T.; Chang, R.C.C.; Wong, G.T.C. The Complement System in the Central Nervous System: From Neurodevelopment to Neurodegeneration. Biomolecules 2022, 12, 337. [Google Scholar] [CrossRef] [PubMed]
- Warwick, C.A.; Keyes, A.L.; Woodruff, T.M.; Usachev, Y.M. The Complement Cascade in the Regulation of Neuroinflammation, Nociceptive Sensitization, and Pain. J. Biol. Chem. 2021, 297, 101085. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Peixoto, C.A.; Oliveira, W.H.d.; Araújo, S.M.d.R.; Nunes, A.K.S. AMPK Activation: Role in the Signaling Pathways of Neuroinflammation and Neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of Glial Nitric Oxide in Neurodegenerative Diseases. Front. Cell Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Fan, W.; Zhu, X.; He, Y.; Zhu, M.; Wu, Z.; Huang, F.; He, H. The Role of Satellite Glial Cells in Orofacial Pain. J. Neurosci. Res. 2019, 97, 393–401. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H.K. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- ten Bosch, G.J.A.; Bolk, J.; ‘t Hart, B.A.; Laman, J.D. Multiple Sclerosis Is Linked to MAPKERK Overactivity in Microglia. J. Mol. Med. 2021, 99, 1033–1042. [Google Scholar] [CrossRef]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 2973. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological Roles of MAPK Signaling Pathways in Human Diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Calvo-Rodriguez, M.; Núñez, L.; Villalobos, C.; Ureña, J.; Guerri, C. Toll-like Receptors in Neuroinflammation, Neurodegeneration, and Alcohol-Induced Brain Damage. IUBMB Life 2021, 73, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Wang, C.; Zepp, J.; Wu, L.; Sun, K.; Zhao, J.; Chandrasekharan, U.; Dicorleto, P.E.; Trapp, B.D.; Ransohoff, R.M.; et al. Act1 Mediates IL-17-Induced EAE Pathogenesis Selectively in NG2+ Glial Cells. Nat. Neurosci. 2013, 16, 1401–1408. [Google Scholar] [CrossRef]
- Lucaciu, A.; Brunkhorst, R.; Pfeilschifter, J.M.; Pfeilschifter, W.; Subburayalu, J. The S1P-S1PR Axis in Neurological Disorders-Insights into Current and Future Therapeutic Perspectives. Cells 2020, 9, 1515. [Google Scholar] [CrossRef]
- Welch, S.P.; Sim-Selley, L.J.; Selley, D.E. Sphingosine-1-Phosphate Receptors as Emerging Targets for Treatment of Pain. Biochem. Pharmacol. 2012, 84, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of Astrocyte Activation by Glycolipids Drives Chronic CNS Inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.Y.; Hoffman, A.S.; Itoh, N.; Ao, Y.; Spence, R.; Sofroniew, M.V.; Voskuhl, R.R. Astrocyte CCL2 Sustains Immune Cell Infiltration in Chronic Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2014, 274, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G. Novel Therapeutic Targets in Neuroinflammation and Neuropathic Pain. Inflamm. Cell Signal 2014, 1, e111. [Google Scholar]
- Allison, R.L.; Ebert, A.D. ALS IPSC-Derived Microglia and Motor Neurons Respond to Astrocyte-Targeted IL-10 and CCL2 Modulation. Hum. Mol. Genet. 2023, 33, 530–542. [Google Scholar] [CrossRef]
- Geng, H.; Chen, L.; Tang, J.; Chen, Y.; Wang, L. The Role of CCL2/CCR2 Axis in Cerebral Ischemia-Reperfusion Injury and Treatment: From Animal Experiments to Clinical Trials. Int. J. Mol. Sci. 2022, 23, 3485. [Google Scholar] [CrossRef]
- Rong, Y.; Ji, C.; Wang, Z.; Ge, X.; Wang, J.; Ye, W.; Tang, P.; Jiang, D.; Fan, J.; Yin, G.; et al. Small Extracellular Vesicles Encapsulating CCL2 from Activated Astrocytes Induce Microglial Activation and Neuronal Apoptosis after Traumatic Spinal Cord Injury. J. Neuroinflamm. 2021, 18, 196. [Google Scholar] [CrossRef]
- Koper, O.M.; Kaminska, J.; Sawicki, K.; Kemona, H. CXCL9, CXCL10, CXCL11, and Their Receptor (CXCR3) in Neuroinflammation and Neurodegeneration. Adv. Clin. Exp. Med. 2018, 27, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.F.; Sha, W.L.; Wu, X.B.; Zhao, L.X.; Ma, L.J.; Gao, Y.J. CXCL10/CXCR3 Signaling in the DRG Exacerbates Neuropathic Pain in Mice. Neurosci. Bull. 2021, 37, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Ransohoff, R.M. Inflammatory Reaction after Traumatic Brain Injury: Therapeutic Potential of Targeting Cell-Cell Communication by Chemokines. Trends Pharmacol. Sci. 2015, 36, 471–480. [Google Scholar] [CrossRef]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-Derived VEGF-A Drives Blood-Brain Barrier Disruption in CNS Inflammatory Disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, E.; Van Damme, P.; Van Den Bosch, L.; Robberecht, W. Vascular Endothelial Growth Factor in Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases. Muscle Nerve 2006, 34, 391–405. [Google Scholar] [CrossRef]
- Zamanian, C.; Kim, G.; Onyedimma, C.; Ghaith, A.K.; Jarrah, R.; Graepel, S.; Moinuddin, F.; Bydon, M. A Review of Vascular Endothelial Growth Factor and Its Potential to Improve Functional Outcomes Following Spinal Cord Injury. Spinal Cord. 2023, 61, 231–237. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely Hominid Features of Adult Human Astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Astillero-Lopez, V.; Villar-Conde, S.; Gonzalez-Rodriguez, M.; Flores-Cuadrado, A.; Ubeda-Banon, I.; Saiz-Sanchez, D.; Martinez-Marcos, A. Proteomic Analysis Identifies HSP90AA1, PTK2B, and ANXA2 in the Human Entorhinal Cortex in Alzheimer’s Disease: Potential Role in Synaptic Homeostasis and Aβ Pathology through Microglial and Astroglial Cells. Brain Pathol. 2024, 22, e13235. [Google Scholar] [CrossRef]
- Soreq, L.; Bird, H.; Mohamed, W.; Hardy, J. Single-Cell RNA Sequencing Analysis of Human Alzheimer’s Disease Brain Samples Reveals Neuronal and Glial Specific Cells Differential Expression. PLoS ONE 2023, 18, e0277630. [Google Scholar] [CrossRef] [PubMed]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-Cell Sequencing of Human Midbrain Reveals Glial Activation and a Parkinson-Specific Neuronal State. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Selvakumar, G.P.; Zaheer, S.; Thangavel, R.; Ahmed, M.E.; Raikwar, S.; Govindarajan, R.; Iyer, S.; Zaheer, A. Cross-Talk between Glia, Neurons and Mast Cells in Neuroinflammation Associated with Parkinson’s Disease. J. Neuroimmune Pharmacol. 2018, 13, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, R.; Kaur, H.; Dubova, I.; Selvakumar, G.P.; Ahmed, M.E.; Raikwar, S.P.; Govindarajan, R.; Kempuraj, D. Parkinson’s Disease Dementia Patients: Expression of Glia Maturation Factor in the Brain. Int. J. Mol. Sci. 2024, 25, 1182. [Google Scholar] [CrossRef]
- Loggia, M.L.; Chonde, D.B.; Akeju, O.; Arabasz, G.; Catana, C.; Edwards, R.R.; Hill, E.; Hsu, S.; Izquierdo-Garcia, D.; Ji, R.R.; et al. Evidence for Brain Glial Activation in Chronic Pain Patients. Brain 2015, 138, 604–615. [Google Scholar] [CrossRef]
- Malpetti, M.; Franzmeier, N.; Brendel, M. PET Imaging to Measure Neuroinflammation In Vivo. Methods Mol. Biol. 2024, 2785, 177–193. [Google Scholar] [PubMed]
- Grace, P.M.; Tawfik, V.L.; Svensson, C.I.; Burton, M.D.; Loggia, M.L.; Hutchinson, M.R. The Neuroimmunology of Chronic Pain: From Rodents to Humans. J. Neurosci. 2021, 41, 855–865. [Google Scholar] [CrossRef]
- Herranz, E.; Giannì, C.; Louapre, C.; Treaba, C.A.; Govindarajan, S.T.; Ouellette, R.; Loggia, M.L.; Sloane, J.A.; Madigan, N.; Izquierdo-Garcia, D.; et al. Neuroinflammatory Component of Gray Matter Pathology in Multiple Sclerosis. Ann. Neurol. 2016, 80, 776–790. [Google Scholar] [CrossRef]
- Lois, C.; González, I.; Izquierdo-García, D.; Zürcher, N.R.; Wilkens, P.; Loggia, M.L.; Hooker, J.M.; Rosas, H.D. Neuroinflammation in Huntington’s Disease: New Insights with 11C-PBR28 PET/MRI. ACS Chem. Neurosci. 2018, 9, 2563–2571. [Google Scholar] [CrossRef]
- Jackson, I.M.; Carlson, M.L.; Beinat, C.; Malik, N.; Kalita, M.; Reyes, S.; Azevedo, E.C.; Nagy, S.C.; Alam, I.S.; Sharma, R.; et al. Clinical Radiosynthesis and Translation of [18F]OP-801: A Novel Radiotracer for Imaging Reactive Microglia and Macrophages. ACS Chem. Neurosci. 2023, 14, 2416–2424. [Google Scholar] [CrossRef]
- Shukuri, M.; Mawatari, A.; Ohno, M.; Suzuki, M.; Doi, H.; Watanabe, Y.; Onoe, H. Detection of Cyclooxygenase-1 in Activated Microglia During Amyloid Plaque Progression: PET Studies in Alzheimer’s Disease Model Mice. J. Nucl. Med. 2016, 57, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, J.; Molotkov, A.; Mintz, A.; Mann, J.J. Progress in PET Imaging of Neuroinflammation Targeting COX-2 Enzyme. Molecules 2021, 26, 3208. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Brückner, M.; Prigent, A.; Sazdovitch, V.; Halle, A.; et al. New Role of P2X7 Receptor in an Alzheimer’s Disease Mouse Model. Mol. Psychiatry 2019, 24, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Daheron, L.; Hurley, H.; Bure, K.; Barker, R.; Carr, A.J.; Williams, D.; Kim, H.W.; French, A.; Coffey, P.J.; et al. Generating IPSCs: Translating Cell Reprogramming Science into Scalable and Robust Biomanufacturing Strategies. Cell Stem Cell 2015, 16, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Albert, K.; Niskanen, J.; Kälvälä, S.; Lehtonen, Š. Utilising Induced Pluripotent Stem Cells in Neurodegenerative Disease Research: Focus on Glia. Int. J. Mol. Sci. 2021, 22, 4334. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.; Han, S.; Kesavamoorthy, G.; Kosugi, M.; Araki, K.; Harada, N.; Kanazawa, M.; Tsukada, H.; Magata, Y.; Ouchi, Y. Differences in in Vitro Microglial Accumulation of the Energy Metabolism Tracers [18F]FDG and [18F]BCPP-EF during LPS- and IL4 Stimulation. Sci. Rep. 2021, 11, 13200. [Google Scholar] [CrossRef] [PubMed]
- Sabogal-Guáqueta, A.M.; Marmolejo-Garza, A.; Trombetta-Lima, M.; Oun, A.; Hunneman, J.; Chen, T.; Koistinaho, J.; Lehtonen, S.; Kortholt, A.; Wolters, J.C.; et al. Species-Specific Metabolic Reprogramming in Human and Mouse Microglia during Inflammatory Pathway Induction. Nat. Commun. 2023, 14, 6454. [Google Scholar] [CrossRef] [PubMed]
- Couch, A.C.M.; Solomon, S.; Duarte, R.R.R.; Marrocu, A.; Sun, Y.; Sichlinger, L.; Matuleviciute, R.; Polit, L.D.; Hanger, B.; Brown, A.; et al. Acute IL-6 Exposure Triggers Canonical IL6Ra Signaling in HiPSC Microglia, but Not Neural Progenitor Cells. Brain Behav. Immun. 2023, 110, 43–59. [Google Scholar] [CrossRef]
- Haukedal, H.; Syshøj Lorenzen, S.; Winther Westi, E.; Corsi, G.I.; Gadekar, V.P.; McQuade, A.; Davtyan, H.; Doncheva, N.T.; Schmid, B.; Chandrasekaran, A.; et al. Alteration of Microglial Metabolism and Inflammatory Profile Contributes to Neurotoxicity in a HiPSC-Derived Microglia Model of Frontotemporal Dementia 3. Brain Behav. Immun. 2023, 113, 353–373. [Google Scholar] [CrossRef]
- Monzón-Sandoval, J.; Burlacu, E.; Agarwal, D.; Handel, A.E.; Wei, L.; Davis, J.; Cowley, S.A.; Cader, M.Z.; Webber, C. Lipopolysaccharide Distinctively Alters Human Microglia Transcriptomes to Resemble Microglia from Alzheimer’s Disease Mouse Models. Dis. Model. Mech. 2022, 15, dmm049349. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; García-Rodríguez, C.; Villalobos, C.; Núñez, L. Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front. Immunol. 2020, 11, 1588. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, E.; Portal, B.; Mothes, T.; Beretta, C.; Lindskog, M.; Erlandsson, A. Intracellular Deposits of Amyloid-Beta Influence the Ability of Human IPSC-Derived Astrocytes to Support Neuronal Function. J. Neuroinflamm. 2023, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Gerasimova, T.; Stepanenko, E.; Novosadova, L.; Arsenyeva, E.; Shimchenko, D.; Tarantul, V.; Grivennikov, I.; Nenasheva, V.; Novosadova, E. Glial Cultures Differentiated from IPSCs of Patients with PARK2-Associated Parkinson’s Disease Demonstrate a Pro-Inflammatory Shift and Reduced Response to TNFα Stimulation. Int. J. Mol. Sci. 2023, 24, 2000. [Google Scholar] [CrossRef] [PubMed]
- Tallantyre, E.C.; Major, P.C.; Atherton, M.J.; Davies, W.A.; Joseph, F.; Tomassini, V.; Pickersgill, T.P.; Harding, K.E.; Willis, M.D.; Winter, M.; et al. How Common Is Truly Benign MS in a UK Population? J. Neurol. Neurosurg. Psychiatry 2019, 90, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Kerkering, J.; Muinjonov, B.; Rosiewicz, K.S.; Diecke, S.; Biese, C.; Schiweck, J.; Chien, C.; Zocholl, D.; Conrad, T.; Paul, F.; et al. IPSC-Derived Reactive Astrocytes from Patients with Multiple Sclerosis Protect Cocultured Neurons in Inflammatory Conditions. J. Clin. Investig. 2023, 133, e164637. [Google Scholar] [CrossRef] [PubMed]
- Marangon, D.; Caporale, N.; Boccazzi, M.; Abbracchio, M.P.; Testa, G.; Lecca, D. Novel in Vitro Experimental Approaches to Study Myelination and Remyelination in the Central Nervous System. Front. Cell Neurosci. 2021, 15, 748849. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Guo, Y.; Biswas, S.; Li, J.; Zhang, H.; Chen, Z.; Deng, W. Promoting Oligodendrocyte Differentiation from Human Induced Pluripotent Stem Cells by Activating Endocannabinoid Signaling for Treating Spinal Cord Injury. Stem Cell Rev. Rep. 2022, 18, 3033–3049. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Bubnys, A.; Tsai, L.H. Harnessing Cerebral Organoids for Alzheimer’s Disease Research. Curr. Opin. Neurobiol. 2022, 72, 120–130. [Google Scholar] [CrossRef]
- Porciúncula, L.O.; Goto-Silva, L.; Ledur, P.F.; Rehen, S.K. The Age of Brain Organoids: Tailoring Cell Identity and Functionality for Normal Brain Development and Disease Modeling. Front. Neurosci. 2021, 15, 674563. [Google Scholar] [CrossRef] [PubMed]
- Sabate-Soler, S.; Nickels, S.L.; Saraiva, C.; Berger, E.; Dubonyte, U.; Barmpa, K.; Lan, Y.J.; Kouno, T.; Jarazo, J.; Robertson, G.; et al. Microglia Integration into Human Midbrain Organoids Leads to Increased Neuronal Maturation and Functionality. Glia 2022, 70, 1267–1288. [Google Scholar] [CrossRef] [PubMed]
- Morales Pantoja, I.E.; Ding, L.; Leite, P.E.C.; Marques, S.A.; Romero, J.C.; Alam El Din, D.M.; Zack, D.J.; Chamling, X.; Smirnova, L. A Novel Approach to Increase Glial Cell Populations in Brain Microphysiological Systems. Adv. Biol. 2023, e2300198. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, L.; Novak, S.W.; Yu, J.; Gallina, I.S.; Xu, L.L.; Lim, C.K.; Fernandes, S.; Shokhirev, M.N.; Williams, A.E.; et al. Morphological Diversification and Functional Maturation of Human Astrocytes in Glia-Enriched Cortical Organoid Transplanted in Mouse Brain. Nat. Biotechnol. 2024. [CrossRef]
- Park, D.S.; Kozaki, T.; Tiwari, S.K.; Moreira, M.; Khalilnezhad, A.; Torta, F.; Olivié, N.; Thiam, C.H.; Liani, O.; Silvin, A.; et al. IPS-Cell-Derived Microglia Promote Brain Organoid Maturation via Cholesterol Transfer. Nature 2023, 623, 397–405. [Google Scholar] [CrossRef]
- Svoboda, D.S.; Barrasa, M.I.; Shu, J.; Rietjens, R.; Zhang, S.; Mitalipova, M.; Berube, P.; Fu, D.; Shultz, L.D.; Bell, G.W.; et al. Human IPSC-Derived Microglia Assume a Primary Microglia-like State after Transplantation into the Neonatal Mouse Brain. Proc. Natl. Acad. Sci. USA 2019, 116, 25293–25303. [Google Scholar] [CrossRef]
- Xu, R.; Li, X.; Boreland, A.J.; Posyton, A.; Kwan, K.; Hart, R.P.; Jiang, P. Human IPSC-Derived Mature Microglia Retain Their Identity and Functionally Integrate in the Chimeric Mouse Brain. Nat. Commun. 2020, 11, 1577. [Google Scholar] [CrossRef]
- Espuny-Camacho, I.; Arranz, A.M.; Fiers, M.; Snellinx, A.; Ando, K.; Munck, S.; Bonnefont, J.; Lambot, L.; Corthout, N.; Omodho, L.; et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 2017, 93, 1066–1081.e8. [Google Scholar] [CrossRef]
- Hasselmann, J.; Coburn, M.A.; England, W.; Figueroa Velez, D.X.; Kiani Shabestari, S.; Tu, C.H.; McQuade, A.; Kolahdouzan, M.; Echeverria, K.; Claes, C.; et al. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 2019, 103, 1016–1033.e10. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, Y.; Xie, Y.; Zhang, L.; Tang, P. Cell Transplantation for Spinal Cord Injury: Tumorigenicity of Induced Pluripotent Stem Cell-Derived Neural Stem/Progenitor Cells. Stem Cells Int. 2018, 2018, 5653787. [Google Scholar] [CrossRef]
- Profico, D.C.; Gelati, M.; Ferrari, D.; Sgaravizzi, G.; Ricciolini, C.; Projetti Pensi, M.; Muzi, G.; Cajola, L.; Copetti, M.; Ciusani, E.; et al. Human Neural Stem Cell-Based Drug Product: Clinical and Nonclinical Characterization. Int. J. Mol. Sci. 2022, 23, 13425. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Liang, F.; Wu, G.; Cao, F.; Liu, J.; He, Z.; Wang, C.; Zhu, L.; Chen, X.; Yang, Y. The Landscape of Biomimetic Nanovesicles in Brain Diseases. Adv. Mater. 2024, 36, e2306583. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tan, X.; Li, S.; Al-Nusaif, M.; Le, W. Role of Glia-Derived Extracellular Vesicles in Neurodegenerative Diseases. Front. Aging Neurosci. 2021, 13, 765395. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, M.; Tozzi, F.; Verderio, C.; Origlia, N. Emerging Roles of Extracellular Vesicles in Alzheimer’s Disease: Focus on Synaptic Dysfunction and Vesicle-Neuron Interaction. Cells 2022, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- López-Guerrero, J.A.; Ripa, I.; Andreu, S.; Bello-Morales, R. The Role of Extracellular Vesicles in Demyelination of the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 9111. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Wang, Y.; Xiao, Y.; Peng, M.; Mai, W.; Hu, B.; Jia, Y.; Chen, H.; Yang, Y.; Xiang, Q.; et al. Extracellular Vesicles Derived from Astrocyte-Treated with HaFGF14-154 Attenuate Alzheimer Phenotype in AD Mice. Theranostics 2022, 12, 3862–3881. [Google Scholar] [CrossRef]
- Raffaele, S.; Gelosa, P.; Bonfanti, E.; Lombardi, M.; Castiglioni, L.; Cimino, M.; Sironi, L.; Abbracchio, M.P.; Verderio, C.; Fumagalli, M. Microglial Vesicles Improve Post-Stroke Recovery by Preventing Immune Cell Senescence and Favoring Oligodendrogenesis. Mol. Ther. 2021, 29, 1439–1458. [Google Scholar] [CrossRef]
- Hering, C.; Shetty, A.K. Extracellular Vesicles Derived From Neural Stem Cells, Astrocytes, and Microglia as Therapeutics for Easing TBI-Induced Brain Dysfunction. Stem Cells Transl. Med. 2023, 12, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Iqbal, Z.; Xu, L.; Wen, C.; Duan, L.; Xia, J.; Yang, N.; Zhang, Y.; Liang, Y. Brain-Derived Extracellular Vesicles: Potential Diagnostic Biomarkers for Central Nervous System Diseases. Psychiatry Clin. Neurosci. 2024, 78, 83–96. [Google Scholar] [CrossRef]
- Vicente, M.C.; Paneghini, J.L.; Stabile, A.M.; Amorim, M.; Anibal Silva, C.E.; Patrone, L.G.A.; Cunha, T.M.; Bícego, K.C.; Almeida, M.C.; Carrettiero, D.C.; et al. Inhibition of Pro-Inflammatory Microglia with Minocycline Improves Cognitive and Sleep-Wake Dysfunction Under Respiratory Stress in a Sporadic Model for Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 95, 317–337. [Google Scholar] [CrossRef]
- Scott, G.; Zetterberg, H.; Jolly, A.; Cole, J.H.; De Simoni, S.; Jenkins, P.O.; Feeney, C.; Owen, D.R.; Lingford-Hughes, A.; Howes, O.; et al. Minocycline Reduces Chronic Microglial Activation after Brain Trauma but Increases Neurodegeneration. Brain 2018, 141, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Pechacek, K.M.; Reck, A.M.; Frankot, M.A.; Vonder Haar, C. Minocycline Fails to Treat Chronic Traumatic Brain Injury-Induced Impulsivity and Attention Deficits. Exp. Neurol. 2022, 348, 113924. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive Multiple Sclerosis: From Pathophysiology to Therapeutic Strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Möller, T.; Bard, F.; Bhattacharya, A.; Biber, K.; Campbell, B.; Dale, E.; Eder, C.; Gan, L.; Garden, G.A.; Hughes, Z.A.; et al. Critical Data-Based Re-Evaluation of Minocycline as a Putative Specific Microglia Inhibitor. Glia 2016, 64, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Kriz, J.; Nguyen, M.D.; Julien, J.P. Minocycline Slows Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2002, 10, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.A.; Kim, T.U.; Chang, M.C. Minocycline for Controlling Neuropathic Pain: A Systematic Narrative Review of Studies in Humans. J. Pain. Res. 2021, 14, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Q.; Liu, D.Q.; Chen, S.P.; Sun, J.; Wang, X.M.; Tian, Y.K.; Wu, W.; Ye, D.W. Minocycline as a Promising Therapeutic Strategy for Chronic Pain. Pharmacol. Res. 2018, 134, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, M.; Li, Y.; Li, Y.; Hua, Y.; Fan, Y. Minocycline Promotes Functional Recovery in Ischemic Stroke by Modulating Microglia Polarization through STAT1/STAT6 Pathways. Biochem. Pharmacol. 2021, 186, 114464. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P.; et al. Minocycline at 2 Different Dosages vs Placebo for Patients With Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef]
- Clarke, A.R.; Christophe, B.R.; Khahera, A.; Sim, J.L.; Connolly, E.S. Therapeutic Modulation of the Complement Cascade in Stroke. Front. Immunol. 2019, 10, 1723. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Ager, R.R.; Chu, S.-H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR Antagonist Decreases Pathology and Enhances Behavioral Performance in Murine Models of Alzheimer’s Disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Sewell, D.L.; Nacewicz, B.; Liu, F.; Macvilay, S.; Erdei, A.; Lambris, J.D.; Sandor, M.; Fabry, Z. Complement C3 and C5 Play Critical Roles in Traumatic Brain Cryoinjury: Blocking Effects on Neutrophil Extravasation by C5a Receptor Antagonist. J. Neuroimmunol. 2004, 155, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative Analysis of Cellular Inflammation after Traumatic Spinal Cord Injury: Evidence for a Multiphasic Inflammatory Response in the Acute to Chronic Environment. Brain 2010, 133, 433–447. [Google Scholar] [CrossRef]
- Lee, J.D.; Kumar, V.; Fung, J.N.T.; Ruitenberg, M.J.; Noakes, P.G.; Woodruff, T.M. Pharmacological Inhibition of Complement C5a-C5a1 Receptor Signalling Ameliorates Disease Pathology in the HSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 2017, 174, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.T.; Ajit, D.; Camden, J.M.; Erb, L.; Weisman, G.A. Purinergic Receptors as Potential Therapeutic Targets in Alzheimer’s Disease. Neuropharmacology 2016, 104, 169–179. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Yang, G. Adenosinergic Pathway in Parkinson’s Disease: Recent Advances and Therapeutic Perspective. Mol. Neurobiol. 2023, 60, 3054–3070. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Zabala, A.; Matute, C. Purinergic Receptors in Multiple Sclerosis Pathogenesis. Brain Res. Bull. 2019, 151, 38–45. [Google Scholar] [CrossRef]
- Oliveira-Giacomelli, Á.; Naaldijk, Y.; Sardá-Arroyo, L.; Gonçalves, M.C.B.; Corrêa-Velloso, J.; Pillat, M.M.; de Souza, H.D.N.; Ulrich, H. Purinergic Receptors in Neurological Diseases With Motor Symptoms: Targets for Therapy. Front. Pharmacol. 2018, 9, 325. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, Z.; Ji, R.; Zhu, J.; Sui, Q.Q.; Knight, G.E.; Burnstock, G.; He, C.; Yuan, H.; Xiang, Z. Inhibition of P2X7 Receptors Improves Outcomes after Traumatic Brain Injury in Rats. Purinergic Signal 2017, 13, 529–544. [Google Scholar] [CrossRef]
- Li, F.; Xu, D.; Hou, K.; Gou, X.; Li, Y. The Role of P2Y12 Receptor Inhibition in Ischemic Stroke on Microglia, Platelets and Vascular Smooth Muscle Cells. J. Thromb. Thrombolysis 2020, 50, 874–885. [Google Scholar] [CrossRef]
- Wen, R.X.; Shen, H.; Huang, S.X.; Wang, L.P.; Li, Z.W.; Peng, P.; Mamtilahun, M.; Tang, Y.H.; Shen, F.X.; Tian, H.L.; et al. P2Y6 Receptor Inhibition Aggravates Ischemic Brain Injury by Reducing Microglial Phagocytosis. CNS Neurosci. Ther. 2020, 26, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Ceruti, S. The Role of Adenosine and P2Y Receptors Expressed by Multiple Cell Types in Pain Transmission. Brain Res. Bull. 2019, 151, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhou, Z.; Sha, W.; Wang, L.; Yan, F.; Yang, X.; Qin, X.; Wu, M.; Li, D.; Tian, S.; et al. A Novel CX3CR1 Inhibitor AZD8797 Facilitates Early Recovery of Rat Acute Spinal Cord Injury by Inhibiting Inflammation and Apoptosis. Int. J. Mol. Med. 2020, 45, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Liang, M.; Zheng, H.; Fan, C.; Zhang, H.; Lu, X.; Du, Z.; Lian, Y.; Zhang, Y.; Bi, X. Anti-Mouse CX3CR1 Antibody Alleviates Cognitive Impairment, Neuronal Loss and Myelin Deficits in an Animal Model of Brain Ischemia. Neuroscience 2020, 438, 169–181. [Google Scholar] [CrossRef]
- Zhao, P.; Xu, Y.; Jiang, L.L.; Fan, X.; Li, L.; Li, X.; Arase, H.; Zhao, Y.; Cao, W.; Zheng, H.; et al. A Tetravalent TREM2 Agonistic Antibody Reduced Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Sci. Transl. Med. 2022, 14, eabq0095. [Google Scholar] [CrossRef] [PubMed]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 Activation on Microglia Promotes Myelin Debris Clearance and Remyelination in a Model of Multiple Sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Y.; Wang, L.; Li, Z.; Tang, S.; Wang, Y.; Gu, N.; Sun, X.; Li, L. TREM2 Activation Alleviates Neural Damage via Akt/CREB/BDNF Signalling after Traumatic Brain Injury in Mice. J. Neuroinflamm. 2022, 19, 289. [Google Scholar] [CrossRef]
- Pérez-Olives, C.; Rivas-Santisteban, R.; Lillo, J.; Navarro, G.; Franco, R. Recent Advances in the Potential of Cannabinoids for Neuroprotection in Alzheimer’s, Parkinson’s, and Huntington’s Diseases. Adv. Exp. Med. Biol. 2021, 1264, 81–92. [Google Scholar]
- Duffy, S.S.; Hayes, J.P.; Fiore, N.T.; Moalem-Taylor, G. The Cannabinoid System and Microglia in Health and Disease. Neuropharmacology 2021, 190, 108555. [Google Scholar] [CrossRef]
- Li, L.; Luo, Q.; Shang, B.; Yang, X.; Zhang, Y.; Pan, Q.; Wu, N.; Tang, W.; Du, D.; Sun, X.; et al. Selective Activation of Cannabinoid Receptor-2 Reduces White Matter Injury via PERK Signaling in a Rat Model of Traumatic Brain Injury. Exp. Neurol. 2022, 347, 113899. [Google Scholar] [CrossRef]
- Vicente-Acosta, A.; Ceprian, M.; Sobrino, P.; Pazos, M.R.; Loría, F. Cannabinoids as Glial Cell Modulators in Ischemic Stroke: Implications for Neuroprotection. Front. Pharmacol. 2022, 13, 888222. [Google Scholar] [CrossRef] [PubMed]
- Hagan, N.; Kane, J.L.; Grover, D.; Woodworth, L.; Madore, C.; Saleh, J.; Sancho, J.; Liu, J.; Li, Y.; Proto, J.; et al. CSF1R Signaling Is a Regulator of Pathogenesis in Progressive MS. Cell Death Dis. 2020, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological Targeting of CSF1R Inhibits Microglial Proliferation and Prevents the Progression of Alzheimer’s-like Pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.L.; Fleming, S.M.; Budge, K.M.; Boyle, A.M.; Kim, C.; Alam, G.; Beier, E.E.; Wu, L.J.; Richardson, J.R. Pharmacological Inhibition of CSF1R by GW2580 Reduces Microglial Proliferation and Is Protective against Neuroinflammation and Dopaminergic Neurodegeneration. FASEB J. 2020, 34, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Muriana, A.; Mancuso, R.; Francos-Quijorna, I.; Olmos-Alonso, A.; Osta, R.; Perry, V.H.; Navarro, X.; Gomez-Nicola, D.; López-Vales, R. CSF1R Blockade Slows the Progression of Amyotrophic Lateral Sclerosis by Reducing Microgliosis and Invasion of Macrophages into Peripheral Nerves. Sci. Rep. 2016, 6, 25663. [Google Scholar] [CrossRef] [PubMed]
- Gerber, Y.N.; Saint-Martin, G.P.; Bringuier, C.M.; Bartolami, S.; Goze-Bac, C.; Noristani, H.N.; Perrin, F.E. CSF1R Inhibition Reduces Microglia Proliferation, Promotes Tissue Preservation and Improves Motor Recovery After Spinal Cord Injury. Front. Cell Neurosci. 2018, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.J.; Feng, S.Y.; Qi, Y.P.; Li, K.; Jin, Z.R.; Jing, H.B.; Liu, L.Y.; Cai, J.; Xing, G.G.; Fu, K.Y. Contribution of Microglial Reaction to Increased Nociceptive Responses in High-Fat-Diet (HFD)-Induced Obesity in Male Mice. Brain Behav. Immun. 2019, 80, 777–792. [Google Scholar] [CrossRef]
- Ritzel, R.M.; He, J.; Li, Y.; Cao, T.; Khan, N.; Shim, B.; Sabirzhanov, B.; Aubrecht, T.; Stoica, B.A.; Faden, A.I.; et al. Proton Extrusion during Oxidative Burst in Microglia Exacerbates Pathological Acidosis Following Traumatic Brain Injury. Glia 2021, 69, 746–764. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xu, Y.; Chen, S.; Fang, M. Inhibited CSF1R Alleviates Ischemia Injury via Inhibition of Microglia M1 Polarization and NLRP3 Pathway. Neural Plast. 2020, 2020, 8825954. [Google Scholar] [CrossRef]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (Fingolimod) Efficacy in an Animal Model of Multiple Sclerosis Requires Astrocyte Sphingosine 1-Phosphate Receptor 1 (S1P1) Modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus Placebo in Secondary Progressive Multiple Sclerosis (EXPAND): A Double-Blind, Randomised, Phase 3 Study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Piperi, C. Beneficial Effects of Fingolimod in Alzheimer’s Disease: Molecular Mechanisms and Therapeutic Potential. Neuromol. Med. 2019, 21, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Doolen, S.; Iannitti, T.; Donahue, R.R.; Shaw, B.C.; Grachen, C.M.; Taylor, B.K. Fingolimod Reduces Neuropathic Pain Behaviors in a Mouse Model of Multiple Sclerosis by a Sphingosine-1 Phosphate Receptor 1-Dependent Inhibition of Central Sensitization in the Dorsal Horn. Pain 2018, 159, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Sood, A.; Jain, R.; Kamatham, P.T.; Khatri, D.K. Fingolimod Exerts Neuroprotection by Regulating S1PR1 Mediated BNIP3-PINK1-Parkin Dependent Mitophagy in Rotenone Induced Mouse Model of Parkinson’s Disease. Neurosci. Lett. 2024, 820, 137596. [Google Scholar] [CrossRef] [PubMed]
- Potenza, R.L.; De Simone, R.; Armida, M.; Mazziotti, V.; Pèzzola, A.; Popoli, P.; Minghetti, L. Fingolimod: A Disease-Modifier Drug in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2016, 13, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.D.; Paganoni, S.; Atassi, N.; Macklin, E.A.; Goyal, N.; Rivner, M.; Simpson, E.; Appel, S.; Grasso, D.L.; Mejia, N.I.; et al. Phase IIa Trial of Fingolimod for Amyotrophic Lateral Sclerosis Demonstrates Acceptable Acute Safety and Tolerability. Muscle Nerve 2017, 56, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Lu, P.; Cai, Y.; Wang, Y.; Hong, L.; Ren, H.; Heng, B.C.; Liu, H.; Zhou, J.; et al. Local Delivery of FTY720 in PCL Membrane Improves SCI Functional Recovery by Reducing Reactive Astrogliosis. Biomaterials 2015, 62, 76–87. [Google Scholar] [CrossRef]
- Cheng, H.; Di, G.; Gao, C.C.; He, G.; Wang, X.; Han, Y.L.; Sun, L.A.; Zhou, M.L.; Jiang, X. FTY720 Reduces Endothelial Cell Apoptosis and Remodels Neurovascular Unit after Experimental Traumatic Brain Injury. Int. J. Med. Sci. 2021, 18, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Naseh, M.; Vatanparast, J.; Rafati, A.; Bayat, M.; Haghani, M. The Emerging Role of FTY720 as a Sphingosine 1-Phosphate Analog for the Treatment of Ischemic Stroke: The Cellular and Molecular Mechanisms. Brain Behav. 2021, 11, e02179. [Google Scholar] [CrossRef]
- Wallin, J.; Svenningsson, P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5606. [Google Scholar] [CrossRef]
- Lai, J.; Mei, Z.L.; Wang, H.; Hu, M.; Long, Y.; Miao, M.X.; Li, N.; Hong, H. Montelukast Rescues Primary Neurons against Aβ1-42-Induced Toxicity through Inhibiting CysLT1R-Mediated NF-ΚB Signaling. Neurochem. Int. 2014, 75, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Biber, N.; Toklu, H.Z.; Solakoglu, S.; Gultomruk, M.; Hakan, T.; Berkman, Z.; Gul Dulger, F. Cysteinyl-Leukotriene Receptor Antagonist Montelukast Decreases Blood-Brain Barrier Permeability but Does Not Prevent Oedema Formation in Traumatic Brain Injury. Brain Inj. 2009, 23, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Shi, X.; Huang, H.; Zhu, Y.; Wu, Y. Montelukast Attenuates Neuropathic Pain through Inhibiting P38 Mitogen-Activated Protein Kinase and Nuclear Factor-Kappa B in a Rat Model of Chronic Constriction Injury. Anesth. Analg. 2014, 118, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Gelosa, P.; Bonfanti, E.; Castiglioni, L.; Delgado-Garcia, J.M.; Gruart, A.; Fontana, L.; Gotti, M.; Tremoli, E.; Lecca, D.; Fumagalli, M.; et al. Improvement of Fiber Connectivity and Functional Recovery after Stroke by Montelukast, an Available and Safe Anti-Asthmatic Drug. Pharmacol. Res. 2019, 142, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Šimončičová, E.; Gonçalves de Andrade, E.; Vecchiarelli, H.A.; Awogbindin, I.O.; Delage, C.I.; Tremblay, M.È. Present and Future of Microglial Pharmacology. Trends Pharmacol. Sci. 2022, 43, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.È. Microglial Functional Alteration and Increased Diversity in the Challenged Brain: Insights into Novel Targets for Intervention. Brain Behav. Immun. Health 2021, 16, 100301. [Google Scholar] [CrossRef]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and Pain: Is Chronic Pain a Gliopathy? Pain 2013, 154 (Suppl. 1), S10–S28. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Signaling Pathway | Functions | Glial Cells | Pathology | Ref |
|---|---|---|---|---|
| Tropomyosine receptor kinase B (TrKB) | Regulates nitric oxide release and supports neuroinflammation. | Astrocytes Microglia | MS Pain | [17,18] |
| Nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) | Its activation and subsequent transcription of pro-inflammatory factors triggers inflammation and neurodegeneration. | Astrocytes Microglia | AD PD MS Pain TBI ALS Ischemic stroke | [19,20,21] |
| JAK/STAT pathway | Regulates homeostasis in inflammatory circumstances, cellular functions that mediate innate and adaptive immunity and cytokine production. | Astrocytes Microglia | AD PD MS Pain SCI Ischemic stroke | [21,22,23,24,25] |
| Purinergic receptors | These are activated by extracellular nucleotides and nucleosides whose extracellular concentrations rise following tissue damage or oxygen deprivation. | Astrocytes Microglia SGCs | AD PD MS Pain ALS Ischemic stroke | [26,27] |
| Neurotransmitters (glutamate, GABA) | These play a critical role in maintaining the excitation–inhibition balance. Alterations in this equilibrium contribute to neurodegeneration. They also modulate the afferent transmission of nociceptive information. | Astrocytes Microglia SGCs | AD PD MS Pain ALS | [28,29,30] |
| CX3CL1/CX3CR1 | Its deficiency is correlated with a worsening of neurodegeneration. | Microglia SGCs | AD PD MS Pain TBI ALS Ischemic stroke | [31] |
| Complement system | It is involved in the control of microglial functions, such as motility, phagocytosis and cytokine release. It protects the brain from pathogens and potentially harmful stimuli, such as aberrant and misfolded proteins. | Astrocytes Microglia | AD PD MS ALS Ischemic stroke | [32,33] |
| Triggering receptor expressed on myeloid cells 2 (TREM2) | Expressed by an activated phenotype of microglia with protective functions for the maintenance of CNS tissue homeostasis, regulation of inflammation and phagocytosis. | Microglia | AD PD MS Pain TBI ALS Ischemic stroke | [34] |
| PI3K/Akt pathway | It is involved in apoptosis and regulation of inflammatory responses. | Microglia | AD PD ALS Pain | [21,35] |
| AMP-activated protein kinase (AMPK) | It maintains steady cellular energy levels by stimulating glucose and fatty acid uptake and oxidation in the event of energy depletion. | Microglia | AD PD MS Pain | [36] |
| Nitric oxide (NO) | Signaling molecule synthetized by enzymes activated only in pathological conditions. | Astrocytes Microglia SGCs | AD PD MS Pain ALS | [37,38] |
| Mitogen-activated protein kinase (MAPK) | Serine/threonine protein kinase with significant roles in cell proliferation, differentiation and apoptosis. | Microglia SGCs | AD PD MS Pain ALS | [21,39,40,41,42] |
| Toll-like receptors (TLRs) | Responsible for persistent neuroinflammation | Astrocytes Microglia | AD PD ALS Ischemic stroke | [43] |
| NFkB activator 1 (Act1) | Triggers the production of pro-inflammatory cytokines, chemokines and metalloproteinases. | Astrocytes | MS | [44] |
| Sphingosine 1-phosphate (S1P1) | Regulates cellular growth, survival and differentiation by binding to specific G-protein-coupled receptors. | Astrocytes Microglia | AD PD MS Pain ALS Ischemic stroke | [45,46] |
| β-1,4-galactosyltransferase 6 (B4GALT6) | It synthesizes lactosylceramide (LacCer), a lipid mediator that triggers inflammation and astrogliosis. | Astrocytes | MS | [47] |
| Chemokine (C-C motif) ligand 2 (CCL2) | Regulates immune cell recruitment to the site of inflammation. | Astrocytes Microglia | MS Pain SCI TBI ALS Ischemic stroke | [21,48,49,50,51,52] |
| C-X-C motif chemokine ligand 10 (CXCL10) | Regulates the recruitment of infiltrating immune cells into CNS lesions during neuroinflammation. | Astrocytes Microglia SGCs | AD MS Pain TBI | [53,54,55] |
| Vascular endothelial growth factor (VEGF) | Supports vascular permeability and CNS damage in acute inflammatory lesions. | Astrocytes | AD PD MS Pain ALS SCI Ischemic stroke | [56,57,58] |
| Pharmacological Agents | Target Glial Cells | Mechanism of Action | Pathology and Species | Ref. |
|---|---|---|---|---|
| Minocycline | Microglia Astrocytes Potential influence on peripheral myeloid cells, oligodendrocytes, neurons, and endothelial cells. | A tetracycline-derived antibiotic with inhibitory effects on microglial pro-inflammatory cytokine release and phagocytosis. | Rodent AD → reduces microglial recruitment and recovers cognitive performance Human AD → no effects on cognitive or functional impairments | [110,111,112,113,114,115,116,117,118,119] |
| Rodent MS → effects on disease course Human MS → no effects on relapses Rodent pain → strong analgesic effect in animal models of chronic pain Human pain → mixed results Rodent ALS → slows disease progression Human ALS → worsens disease progression Rodent TBI → no effects Human TBI → mixed results Rodent ischemic stroke → promotes functional recovery by modulating microglia polarization | ||||
| Complement pathway inhibitors | Microglia | Antagonists of elements of the complement cascade, they modulate microglial state and interactions with synapses. | Rodent AD → C5aR1 antagonists reduce cognitive decline and attenuate microglial activation Rodent ALS → C5aR1 antagonists slow disease progression Human ALS → humanized C1q antibody in Phase II clinical trial Rodent TBI → C5aR1 inhibitors reduce pathology severity Rodent SCI → early administration of C5aR1 antagonists accelerate recovery Rodent ischemic stroke → phase-specific C3-blocking antibodies reduce acute injury extent | [120,121,122,123,124] |
| Purinergic receptors modulators | Microglia Astrocytes Satellite glial cells Effects on oligodendrocytes and neurons and on various cell types (e.g., antiaggregating effect on platelets by marketed thienopyridine and other P2Y12 antagonists). | Agonists and antagonists of several purinergic receptors that are involved in CNS and PNS disorders. | Rodent AD → reduced neuroinflammation and neurotoxicity Rodent PD → antagonist of A2A, P2X1, P2X7 and P2Y1 receptor subtypes decrease microglia activation and slow down disease progression Rodent MS → activation or blockade of P2X4, P2X7 and P2Y12 modify disease course Rodent pain → antagonists at P2X and P2Y and agonists at A3 receptor subtypes have positive effects on different pain types Rodent ALS → antagonism of P2X7 may be beneficial at late pre-symptomatic stages Rodent TBI → Inhibition of P2X7 improves pathology outcomes, reducing microglial activation Rodent ischemic stroke → P2Y12 antagonists exert neuroprotective and anti-inflammatory effects. Inhibition of microglial phagocytosis by selective P2Y6 inhibitor aggravates neurological functions. | [125,126,127,128,129,130,131,132] |
| Fractalkine signaling inhibitors | Microglia Influence on peripheral myeloid cells and oligodendrocyte precursor cells. | Antagonists of CX3CR1, they act on various microglial functions (i.e., modulation of neurotransmission, neurotrophic support and regulation of inflammatory response.) | Rodent SCI → CX3CR1 inhibitors facilitate early recovery Rodent ischemic stroke → CX3CR1 antibody alleviates cognitive impairment, neuronal loss and myelin deficits | [133,134] |
| TREM2 agonists | Microglia Influence on peripheral myeloid cells. | They act on a receptor of the immunoglobulin superfamily that regulates microglial survival, proliferation, phagocytosis and metabolic state. | Rodent AD → enhance microglia functions and reduce amyloid pathology Human AD → Phase II and III clinical trials Rodent MS → accelerate myelin debris removal by microglia Rodent TBI → alleviate neural damage | [135,136,137] |
| Cannabinoids | Microglia Astrocytes | Agonists at cannabinoid receptors CB1R and CB2R, whose activation reduces pro-inflammatory cytokine production and promotes cell migration. | Rodent PD → reduction of glial activation and protection of dopaminergic neurons Rodent AD → reduction of oxidative stress and neuroinflammation Rodent MS → reduction of the clinical severity of the pathology and decrease of microglia activation Rodent pain → non-selective CB1/2 agonist reduces neuropathic pain and microglial activation Rodent ALS → CB2R agonist improves motor function and reduces microglial activation Rodent TBI → selective CB2 agonist protects white matter and drives microglial polarization toward a protective phenotype Rodent ischemic stroke → controversial results | [138,139,140,141] |
| Colony stimulating factor 1 receptor (CSF1R) inhibitors | Microglia Potential effects on astrocytes and peripheral immune cells. | They act on a receptor tyrosine kinase required for the development, maintenance and proliferation of microglia. | Rodent AD → inhibition of microglial proliferation and prevention of disease progression Rodent PD → reduction of microglial proliferation and protection against neuroinflammation and dopaminergic neurodegeneration Rodent MS → attenuation of microglial activation, blockade of axonal damage and neurological impairments Rodent pain → elimination of microglia and reduction of inflammation Rodent ALS → slow down disease progression by reducing microgliosis Human ALS → in Phase II and III clinical trials Rodent SCI → reduction of microglial proliferation and improvement of motor recovery Rodent TBI → microglia depletion and decreased inflammation Rodent ischemic stroke → neuroprotective effect by inhibiting microglia polarization | [142,143,144,145,146,147,148,149] |
| S1PR inhibitors (fingolimod, siponimod) | Astrocytes General effects on immune cells (i.e., in MS they maintain lymphocytes within lymph nodes thus limiting penetration in the CNS). Already on the market as first oral therapy for MS. | They inhibit the inflammatory responses in the brain by acting on S1PRs, principally S1PR1, and are involved in multiple processes including cell survival, proliferation, differentiation and migration. | Rodent AD → beneficial effects on AD progression by regulating neuroinflammation Animal PD → neuroprotective effect Rodent MS → reduced astrogliosis, demyelination and axonal loss, and improved pathology Human MS → Fingolimod: approved immunosuppressive therapy for RRMS Siponimod: efficacy in Phase III clinical trial Rodent pain → antinociceptive effects in multiple models of peripheral inflammation/injury Rodent ALS → protective and beneficial effects accompanied by a modulation of microglial activation and innate immunity Human ALS → Phase IIa clinical trial Rodent SCI → improved functional recovery by reducing reactive astrogliosis Rodent TBI → attenuation of glia activation Rodent ischemic stroke → reduced lesion size and improved neurological function, decreasing glia activation Human ischemic stroke → effects on a pilot clinical trial | [150,151,152,153,154,155,156,157,158,159] |
| B4GALT5/6 inhibitors | Astrocytes | They inhibit the synthesis of lactosylceramide (LacCer), which in astrocytes acts in an autocrine way, triggering a transcriptional program that promotes neurodegeneration and controls the recruitment and activation of microglia. | Rodent MS → suppress CNS innate immunity and neurodegeneration and interfere with astrocyte activation | [47] |
| Montelukast | Microglia | Leukotriene receptor antagonist, already on the market for asthmatic patients. | Rodent AD → effect on β-amyloid-induced neurotoxicity with a reduction of pro-inflammatory factors Human AD → two ongoing phase II placebo-controlled clinical trials Rodent PD → attenuation of microglial activation and protective effect on motor function deterioration Human PD → ongoing Phase II unblinded clinical study Rodent pain → attenuates neuropathic pain Rodent TBI → attenuates chronic neurological damage caused by neuroinflammation Rodent ischemic stroke → influences microglia phenotype and improves functional recovery | [160,161,162,163,164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magni, G.; Riboldi, B.; Ceruti, S. Human Glial Cells as Innovative Targets for the Therapy of Central Nervous System Pathologies. Cells 2024, 13, 606. https://doi.org/10.3390/cells13070606
Magni G, Riboldi B, Ceruti S. Human Glial Cells as Innovative Targets for the Therapy of Central Nervous System Pathologies. Cells. 2024; 13(7):606. https://doi.org/10.3390/cells13070606
Chicago/Turabian StyleMagni, Giulia, Benedetta Riboldi, and Stefania Ceruti. 2024. "Human Glial Cells as Innovative Targets for the Therapy of Central Nervous System Pathologies" Cells 13, no. 7: 606. https://doi.org/10.3390/cells13070606
APA StyleMagni, G., Riboldi, B., & Ceruti, S. (2024). Human Glial Cells as Innovative Targets for the Therapy of Central Nervous System Pathologies. Cells, 13(7), 606. https://doi.org/10.3390/cells13070606