
The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases



Abstract
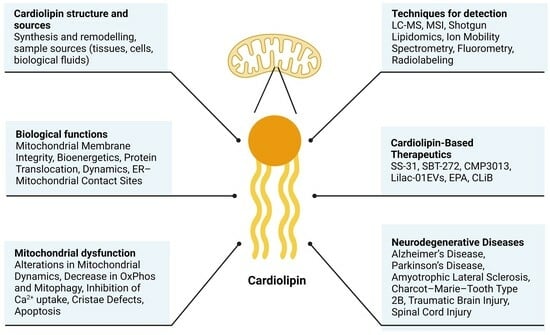
:
1. Introduction
2. Physicochemical Properties and Detection of CL
3. The Role of Cardiolipin in Mitochondrial Biological Functions
3.1. The Role of Cardiolipin in Mitochondrial Membrane Integrity
3.2. Role of Cardiolipin in Bioenergetics
3.3. Role of Cardiolipin in Mitochondrial Protein Translocation
3.4. Role of Cardiolipin in Mitochondrial Dynamics
3.5. Role of Cardiolipin in ER–Mitochondrial Contact Sites
3.6. Other Functions
4. The Role of Cardiolipin Alterations in Mitochondrial Dysfunction
5. The Role of Cardiolipin in Neurodegenerative Diseases
5.1. Alzheimer’s Disease (AD)
5.2. Parkinson’s Disease (PD)
5.3. Amyotrophic Lateral Sclerosis (ALS)
5.4. Charcot–Marie–Tooth Type 2B (CMT2B)
5.5. Traumatic Brain Injury
5.6. Spinal Cord Injury
6. Cardiolipin-Based Therapeutics
7. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Pangborn, M.C. Isolation and Purification of a Serologically Active Phospholipid from Beef Heart. J. Biol. Chem. 1942, 143, 247–256. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of Mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Beltrán-Heredia, E.; Tsai, F.C.; Salinas-Almaguer, S.; Cao, F.J.; Bassereau, P.; Monroy, F. Membrane Curvature Induces Cardiolipin Sorting. Commun. Biol. 2019, 2, 225. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Itoh, K.; Yamada, T.; Cerveny, K.L.; Suzuki, T.L.; Macdonald, P.; Frohman, M.A.; Ramachandran, R.; Iijima, M.; Sesaki, H. Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Mol. Cell 2016, 63, 1034–1043. [Google Scholar] [CrossRef]
- Taylor, W.A.; Hatch, G.M. Identification of the Human Mitochondrial Linoleoyl-Coenzyme a Monolysocardiolipin Acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371. [Google Scholar] [CrossRef]
- Oemer, G.; Lackner, K.; Muigg, K.; Krumschnabel, G.; Watschinger, K.; Sailer, S.; Lindner, H.; Gnaiger, E.; Wortmann, S.B.; Werner, E.R.; et al. Molecular Structural Diversity of Mitochondrial Cardiolipins. Proc. Natl. Acad. Sci. USA 2018, 115, 4158–4163. [Google Scholar] [CrossRef]
- Oemer, G.; Koch, J.; Wohlfarter, Y.; Alam, M.T.; Lackner, K.; Sailer, S.; Neumann, L.; Lindner, H.H.; Watschinger, K.; Haltmeier, M.; et al. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020, 30, 4281–4291.e4. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Trinh, D.; Israwi, A.R.; Arathoon, L.R.; Gleave, J.A.; Nash, J.E. The Multi-Faceted Role of Mitochondria in the Pathology of Parkinson’s Disease. J. Neurochem. 2020, 156, 715–752. [Google Scholar] [CrossRef]
- Aufschnaiter, A.; Kohler, V.; Diessl, J.; Peselj, C.; Carmona-Gutierrez, D.; Keller, W.; Büttner, S. Mitochondrial Lipids in Neurodegeneration. Cell Tissue Res. 2017, 367, 125–140. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Oliveira, M.M.; Melo, T.; Domingues, M.R.M.; Moreira, P.I.; Ferreiro, E.; Peixoto, F.; Videira, R.A. Cardiolipin Profile Changes Are Associated to the Early Synaptic Mitochondrial Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, A.; Garcia-Rozas, P.; Casarejos, M.J.; Pastor, O.; Rodriguez-Navarro, J.A. Lipidomic Alterations in the Mitochondria of Aged Parkin Null Mice Relevant to Autophagy. Front. Neurosci. 2019, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Burg, T.; Rossaert, E.; Moisse, M.; Van Damme, P.; Van Den Bosch, L. Histone Deacetylase Inhibition Regulates Lipid Homeostasis in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 11224. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in Lipid Metabolism of Spinal Cord Linked to Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Hier, Z.E.; Clark, R.S.B.; Kochanek, P.M.; Kagan, V.E.; Bayır, H. Detection of Brain Specific Cardiolipins in Plasma after Experimental Pediatric Head Injury. Exp. Neurol. 2019, 316, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Polimova, A.M.; Maciel, E.; Tyurin, V.A.; Kapralova, V.I.; Winnica, D.E.; Vikulina, A.S.; Domingues, M.R.M.; McCoy, J.; Sanders, L.H.; et al. LC/MS Analysis of Cardiolipins in Substantia Nigra and Plasma of Rotenone-Treated Rats: Implication for Mitochondrial Dysfunction in Parkinson’s Disease. Free Radic. Res. 2015, 49, 681–691. [Google Scholar] [CrossRef]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Lewis, R.N.A.H.; McElhaney, R.N. The Physicochemical Properties of Cardiolipin Bilayers and Cardiolipin-Containing Lipid Membranes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2069–2079. [Google Scholar] [CrossRef]
- Boyd, K.J.; Alder, N.N.; May, E.R. Buckling under Pressure: Curvature-Based Lipid Segregation and Stability Modulation in Cardiolipin-Containing Bilayers. Langmuir 2017, 33, 6937–6946. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a Mitochondrial Cochaperone Associated with Cardiomyopathy, Forms a Complex with Prohibitins to Regulate Cardiolipin Remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevs, P.; Dimitrijevs, P.; Domracheva, I.; Arsenyan, P. Improved Method for the Preparation of Nonyl Acridine Orange Analogues and Utilization in Detection of Cardiolipin. New J. Chem. 2020, 44, 9626–9633. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin Exposure on the Outer Mitochondrial Membrane Modulates α-Synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Cheng, I.F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bömeke, K.; Hübscher, D.; Vukotic, M.; Wanders, R.J.A.; Rehling, P.; Guan, K. Cardiolipin Deficiency Affects Respiratory Chain Function and Organization in an Induced Pluripotent Stem Cell Model of Barth Syndrome. Stem Cell Res. 2013, 11, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.; Ramanathan, A.; Lopez, C.F. Cardiolipin-Dependent Properties of Model Mitochondrial Membranes from Molecular Simulations. Biophys. J. 2019, 117, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.L.; Derreumaux, P.; Nguyen, P.H. Structure and Elasticity of Mitochondrial Membranes: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2023, 127, 10778–10791. [Google Scholar] [CrossRef] [PubMed]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Cristae Membrane Dynamics—A Paradigm Change. Trends Cell Biol. 2020, 30, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organization of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Mourier, A.; Yamada, J.; Michael McCaffery, J.; Nunnari, J. MICOS Coordinates with Respiratory Complexes and Lipids to Establish Mitochondrial Inner Membrane Architecture. eLife 2015, 2015, e07739. [Google Scholar] [CrossRef]
- Rampelt, H.; Wollweber, F.; Gerke, C.; de Boer, R.; van der Klei, I.J.; Bohnert, M.; Pfanner, N.; van der Laan, M. Assembly of the Mitochondrial Cristae Organizer Mic10 Is Regulated by Mic26–Mic27 Antagonism and Cardiolipin. J. Mol. Biol. 2018, 430, 1883–1890. [Google Scholar] [CrossRef]
- Koob, S.; Barrera, M.; Anand, R.; Reichert, A.S. The Non-Glycosylated Isoform of MIC26 Is a Constituent of the Mammalian MICOS Complex and Promotes Formation of Crista Junctions. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Kondadi, A.K.; Meisterknecht, J.; Golombek, M.; Nortmann, O.; Riedel, J.; Peifer-Weiß, L.; Brocke-Ahmadinejad, N.; Schlütermann, D.; Stork, B.; et al. MIC26 and MIC27 Cooperate to Regulate Cardiolipin Levels and the Landscape of OXPHOS Complexes. Life Sci. Alliance 2020, 3, e202000711. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.L.; Robinson, A.J.; Walker, J.E. Cardiolipin Binds Selectively but Transiently to Conserved Lysine Residues in the Rotor of Metazoan ATP Synthases. Proc. Natl. Acad. Sci. USA 2016, 113, 8687–8692. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the Dimeric ATP Synthase from Bovine Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.D.; Ball, W.B.; Pryce, E.N.; Gohil, V.M. Specific Requirements of Nonbilayer Phospholipids in Mitochondrial Respiratory Chain Function and Formation. Mol. Biol. Cell 2016, 27, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The Architecture of Respiratory Supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The Multiple Functions of Cytochrome c and Their Regulation in Life and Death Decisions of the Mammalian Cell: From Respiration to Apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Gao, J.; Siira, S.J.; Shearwood, A.M.; Ermer, J.A.; Hofferek, V.; Mathews, J.C.; Zheng, M.; Reid, G.E.; Rackham, O.; et al. Cardiolipin Is Required for Membrane Docking of Mitochondrial Ribosomes and Protein Synthesis. J. Cell Sci. 2020, 133, jcs240374. [Google Scholar] [CrossRef] [PubMed]
- Senoo, N.; Kandasamy, S.; Ogunbona, O.B.; Baile, M.G.; Lu, Y.; Claypool, S.M. Cardiolipin, Conformation, and Respiratory Complex-Dependent Oligomerization of the Major Mitochondrial ADP/ATP Carrier in Yeast. Sci. Adv. 2020, 6, eabb0780. [Google Scholar] [CrossRef]
- Yi, Q.; Yao, S.; Ma, B.; Cang, X. The Effects of Cardiolipin on the Structural Dynamics of the Mitochondrial ADP/ATP Carrier in Its Cytosol-Open State. J. Lipid Res. 2022, 63, 100227. [Google Scholar] [CrossRef]
- Ghosh, S.; Zulkifli, M.; Joshi, A.; Venkatesan, M.; Cristel, A.; Vishnu, N.; Madesh, M.; Gohil, V.M. MCU-Complex-Mediated Mitochondrial Calcium Signaling Is Impaired in Barth Syndrome. Hum. Mol. Genet. 2022, 31, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ball, W.B.; Madaris, T.R.; Srikantan, S.; Madesh, M.; Mootha, V.K.; Gohil, V.M. An Essential Role for Cardiolipin in the Stability and Function of the Mitochondrial Calcium Uniporter. Proc. Natl. Acad. Sci. USA 2020, 117, 16383–16390. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of Intact Human MCU Supercomplex with the Auxiliary MICU Subunits. Protein Cell 2021, 12, 220–229. [Google Scholar] [CrossRef]
- Li, Y.; Lou, W.; Raja, V.; Denis, S.; Yu, W.; Schmidtke, M.W.; Reynolds, C.A.; Schlame, M.; Houtkooper, R.H.; Greenberg, M.L. Cardiolipin-Induced Activation of Pyruvate Dehydrogenase Links Mitochondrial Lipid Biosynthesis to TCA Cycle Function. J. Biol. Chem. 2019, 294, 11568–11578. [Google Scholar] [CrossRef]
- Li, Y.; Lou, W.; Grevel, A.; Böttinger, L.; Liang, Z.; Ji, J.; Patil, V.A.; Liu, J.; Ye, C.; Hüttemann, M.; et al. Cardiolipin-Deficient Cells Have Decreased Levels of the Iron-Sulfur Biogenesis Protein Frataxin. J. Biol. Chem. 2020, 295, 11928–11937. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, M.; Bharambe, N.; Shang, Y.; Lu, B.; Mandal, A.; Mohan, P.M.; Wang, R.; Boatz, J.C.; Galvez, J.M.M.; Shnyrova, A.V.; et al. NMR Identification of a Conserved Drp1 Cardiolipin-Binding Motif Essential for Stress-Induced Mitochondrial Fission. Proc. Natl. Acad. Sci. USA 2021, 118, e2023079118. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Boopathy, S.; Nguyen, T.H.; Lugo, C.M.; Chao, L.H. Absence of Cardiolipin from the Outer Leaflet of a Mitochondrial Inner Membrane Mimic Restricts Opa1-Mediated Fusion. Front. Mol. Biosci. 2021, 8, 769135. [Google Scholar] [CrossRef] [PubMed]
- von der Malsburg, A.; Sapp, G.M.; Zuccaro, K.E.; von Appen, A.; Moss, F.R.; Kalia, R.; Bennett, J.A.; Abriata, L.A.; Dal Peraro, M.; van der Laan, M.; et al. Structural Mechanism of Mitochondrial Membrane Remodelling by Human OPA1. Nature 2023, 620, 1101–1108. [Google Scholar] [CrossRef]
- Vance, J.E. Inter-Organelle Membrane Contact Sites: Implications for Lipid Metabolism. Biol. Direct 2020, 15, 24. [Google Scholar] [CrossRef]
- Favero, G.; Garcia-Gomez, R.; Monsalve, M.; Rezzani, R.; Lavazza, A.; Stacchiotti, A. Perspective: Mitochondria-ER Contacts in Metabolic Cellular Stress Assessed by Microscopy. Cells 2018, 8, 5. [Google Scholar] [CrossRef]
- Yeo, H.K.; Park, T.H.; Kim, H.Y.; Jang, H.; Lee, J.; Hwang, G.; Ryu, S.E.; Park, S.H.; Song, H.K.; Ban, H.S.; et al. Phospholipid Transfer Function of PTPIP51 at Mitochondria-associated ER Membranes. EMBO Rep. 2021, 22, e51323. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin Remodeling by ALCAT1 Links Oxidative Stress and Mitochondrial Dysfunction to Obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Tirrell, P.S.; Nguyen, K.N.; Luby-Phelps, K.; Friedman, J.R. MICOS Subcomplexes Assemble Independently on the Mitochondrial Inner Membrane in Proximity to ER Contact Sites. J. Cell Biol. 2020, 219, e202003024. [Google Scholar] [CrossRef]
- Iwamoto, M.; Morito, M.; Oiki, S.; Nishitani, Y.; Yamamoto, D.; Matsumori, N. Cardiolipin Binding Enhances KcsA Channel Gating via Both Its Specific and Dianion-Monoanion Interchangeable Sites. iScience 2023, 26, 108471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lyu, J.; Zhu, Y.; Laganowsky, A. Cardiolipin Regulates the Activity of the Mitochondrial ABC Transporter ABCB10. Biochemistry 2023, 62, 3159–3165. [Google Scholar] [CrossRef]
- Liu, G.Y.; Ho Moon, S.; Jenkins, C.M.; Li, M.; Sims, H.F.; Guan, S.; Gross, R.W. The Phospholipase IPLA2 Is a Major Mediator Releasing Oxidized Aliphatic Chains from Cardiolipin, Integrating Mitochondrial Bioenergetics and Signaling. J. Biol. Chem. 2017, 292, 10672–10684. [Google Scholar] [CrossRef]
- Olivar-Villanueva, M.; Ren, M.; Schlame, M.; Phoon, C.K.L. The Critical Role of Cardiolipin in Metazoan Differentiation, Development, and Maturation. Dev. Dyn. 2023, 252, 691–712. [Google Scholar] [CrossRef]
- Schlame, M.; Xu, Y. The Function of Tafazzin, a Mitochondrial Phospholipid–Lysophospholipid Acyltransferase. J. Mol. Biol. 2020, 432, 5043–5051. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov, S.I.; Mulkidjanian, A.Y. Cardiolipin, Perhydroxyl Radicals, and Lipid Peroxidation in Mitochondrial Dysfunctions and Aging. Oxid. Med. Cell. Longev. 2020, 2020, 1323028. [Google Scholar] [CrossRef]
- Vladimirov, G.K.; Vikulina, A.S.; Volodkin, D.; Vladimirov, Y.A. Structure of the Complex of Cytochrome c with Cardiolipin in Non-Polar Environment. Chem. Phys. Lipids 2018, 214, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Belikova, N.A.; Vladimirov, Y.A.; Osipov, A.N.; Kapralov, A.A.; Tyurin, V.A.; Potapovich, M.V.; Basova, L.V.; Peterson, J.; Kurnikov, I.V.; Kagan, V.E. Peroxidase Activity and Structural Transitions of Cytochrome c Bound to Cardiolipin-Containing Membranes. Biochemistry 2006, 45, 4998–5009. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef]
- Vähäheikkilä, M.; Peltomaa, T.; Róg, T.; Vazdar, M.; Pöyry, S.; Vattulainen, I. How Cardiolipin Peroxidation Alters the Properties of the Inner Mitochondrial Membrane? Chem. Phys. Lipids 2018, 214, 15–23. [Google Scholar] [CrossRef]
- Lacombe, M.L.; Tokarska-Schlattner, M.; Boissan, M.; Schlattner, U. The Mitochondrial Nucleoside Diphosphate Kinase (NDPK-D/NME4), a Moonlighting Protein for Cell Homeostasis. Lab. Investig. 2018, 98, 582–588. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination Signal for Mitophagy in Neuronal Cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Iriondo, M.N.; Etxaniz, A.; Varela, Y.R.; Ballesteros, U.; Hervás, J.H.; Montes, L.R.; Goñi, F.M.; Alonso, A. LC3 Subfamily in Cardiolipin-Mediated Mitophagy: A Comparison of the LC3A, LC3B and LC3C Homologs. Autophagy 2022, 18, 2985–3003. [Google Scholar] [CrossRef]
- Clifton, L.A.; Wacklin-Knecht, H.P.; Ådén, J.; Ul Mushtaq, A.; Sparrman, T.; Gröbner, G. Creation of Distinctive Bax-Lipid Complexes at Mitochondrial Membrane Surfaces Drives Pore Formation to Initiate Apoptosis. Sci. Adv. 2023, 9, eadg7940. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Kim, A.A.; Yaguzhinsky, L.S.; Dagda, R.K. Non-Bilayer Structures in Mitochondrial Membranes Regulate ATP Synthase Activity. Physiol. Behav. 2016, 176, 139–148. [Google Scholar] [CrossRef]
- Huang, Y.; Powers, C.; Madala, S.K.; Greis, K.D.; Haffey, W.D.; Towbin, J.A.; Purevjav, E.; Javadov, S.; Strauss, A.W.; Khuchua, Z. Cardiac Metabolic Pathways Affected in the Mouse Model of Barth Syndrome. PLoS ONE 2015, 10, e0128561. [Google Scholar] [CrossRef]
- Anzmann, A.F.; Sniezek, O.L.; Pado, A.; Busa, V.; Vaz, F.M.; Kreimer, S.D.; DeVine, L.R.; Cole, R.N.; Le, A.; Kirsch, B.J.; et al. Diverse Mitochondrial Abnormalities in a New Cellular Model of TAFFAZZIN Deficiency Are Remediated by Cardiolipin-Interacting Small Molecules. J. Biol. Chem. 2021, 297, 101005. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Hatch, A.L.; Higgs, H.N. Effects of Phosphorylation on Drp1 Activation by Its Receptors, Actin, and Cardiolipin. Prepint 2023, 35, ar16. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome C Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Amoscato, A.A.; Sparvero, L.J.; He, R.R.; Watkins, S.; Bayir, H.; Kagan, V.E. Imaging Mass Spectrometry of Diversified Cardiolipin Molecular Species in the Brain. Anal. Chem. 2014, 86, 6587–6595. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Kline, A.E.; Amoscato, A.; Arias, A.S.; Sparvero, L.J.; Tyurin, V.A.; Tyurina, Y.Y.; Fink, B.; Manole, M.D.; Puccio, A.M.; et al. Global Lipidomics Identifies Cardiolipin Oxidation as a Mitochondrial Target for Redox Therapy of Acute Brain Injury. Physiol. Behav. 2012, 176, 139–148. [Google Scholar]
- Schlame, M.; Brody, S.; Hostetler, K.Y. Mitochondrial Cardiolipin in Diverse Eukaryotes: Comparison of Biosynthetic Reactions and Molecular Acyl Species. Eur. J. Biochem. 1993, 212, 727–733. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Amoscato, A.A.; Fink, A.B.; Anthonymuthu, T.; New, L.A.; Kochanek, P.M.; Watkins, S.; Kagan, V.E.; Bayır, H. Imaging Mass Spectrometry Reveals Loss of Polyunsaturated Cardiolipins in the Cortical Contusion, Hippocampus, and Thalamus after Traumatic Brain Injury. J. Neurochem. 2016, 139, 659–675. [Google Scholar] [CrossRef]
- Kolomiytseva, I.K.; Markevich, L.N.; Ignat’Ev, D.A.; Bykova, O.V. Lipids of Nuclear Fractions from Neurons and Glia of Rat Neocortex under Conditions of Artificial Hypobiosis. Biochemistry 2010, 75, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, F.M.; Cafagna, F.; Petruzzella, V.; Gadaleta, M.N.; Quagliariello, E. Lipid Composition in Synaptic and Nonsynaptic Mitochondria from Rat Brains and Effect of Aging. J. Neurochem. 1992, 59, 487–491. [Google Scholar] [CrossRef]
- Kurokin, I.; Lauer, A.A.; Janitschke, D.; Winkler, J.; Theiss, E.L.; Griebsch, L.V.; Pilz, S.M.; Matschke, V.; van der Laan, M.; Grimm, H.S.; et al. Targeted Lipidomics of Mitochondria in a Cellular Alzheimer’s Disease Model. Biomedicines 2021, 9, 81. [Google Scholar] [CrossRef]
- Guan, Z.; Wang, Y.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and Structural Modifications of Phosphatidylethanolamine Plasmalogen in the Brain with Alzheimer Disease. J. Neurophatology Exp. Neurol. 1994, 58, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, A.; Ghio, S.; Caruana, M.; Weckbecker, D.; Schmidt, F.; Kamp, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Giese, A.; et al. Tau-Induced Mitochondrial Membrane Perturbation Is Dependent upon Cardiolipin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183064. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.J.; Ranger, A.L.; Murray, T.; McRae, S.; Klegeris, A. Extracellular Cardiolipin Modulates Glial Phagocytosis and Cytokine Secretion in a Toll-like Receptor 4-dependent Manner. Alzheimer’s Dement. 2020, 16, e047338. [Google Scholar] [CrossRef]
- Kawatani, K.; Holm, M.L.; Starling, S.C.; Martens, Y.A.; Zhao, J.; Lu, W.; Ren, Y.; Li, Z.; Jiang, P.; Jiang, Y.; et al. ABCA7 Deficiency Causes Neuronal Dysregulation by Altering Mitochondrial Lipid Metabolism. Mol. Psychiatry 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.L.; Annesley, S.J.; Gordon, S.E.; Mroczek, K.; Perugini, M.A.; Lawson, V.A.; Fisher, P.R.; Finkelstein, D.I.; Hill, A.F. Misfolded α-Synuclein Causes Hyperactive Respiration without Functional Deficit in Live Neuroblastoma Cells. DMM Dis. Model. Mech. 2020, 13, dmm040899. [Google Scholar] [CrossRef] [PubMed]
- Ghio, S.; Camilleri, A.; Caruana, M.; Ruf, V.C.; Schmidt, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Cauchi, R.J.; Kamp, F.; et al. Cardiolipin Promotes Pore-Forming Activity of Alpha-Synuclein Oligomers in Mitochondrial Membranes. ACS Chem. Neurosci. 2019, 10, 3815–3829. [Google Scholar] [CrossRef] [PubMed]
- Zhaliazka, K.; Ali, A.; Kurouski, D. Phospholipids and Cholesterol Determine Molecular Mechanisms of Cytotoxicity of α-Synuclein Oligomers and Fibrils. ACS Chem. Neurosci. 2023, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, J.; Qi, S.; Liu, Z.; Zhang, X.; Zheng, Y.; Andersen, J.P.; Zhang, W.; Strong, R.; Martinez, P.A.; et al. Cardiolipin Remodeling by ALCAT1 Links Mitochondrial Dysfunction to Parkinson’s Diseases. Aging Cell 2019, 18, e12941. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10, 1759091418781889. [Google Scholar] [CrossRef]
- Phan, K.; He, Y.; Bhatia, S.; Pickford, R.; Mcdonald, G.; Mazumder, S.; Timmins, H.C.; Hodges, J.R.; Piguet, O.; Dzamko, N.; et al. Multiple Pathways of Lipid Dysregulation in Amyotrophic Lateral Sclerosis. Brain Commun. 2023, 5, fcac340. [Google Scholar] [CrossRef]
- Giudetti, A.M.; Guerra, F.; Longo, S.; Beli, R.; Romano, R.; Manganelli, F.; Nolano, M.; Mangini, V.; Santoro, L.; Bucci, C. An Altered Lipid Metabolism Characterizes Charcot-Marie-Tooth Type 2B Peripheral Neuropathy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158805. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Lin, C.; Zuo, Q.; Liu, Y.; Xiao, M.; Xu, X.; Li, Z.; Bao, Z.; Chen, H.; You, Y.; et al. Cardiolipin-Dependent Mitophagy Guides Outcome after Traumatic Brain Injury. J. Neurosci. 2019, 39, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.K.; Deng, L.X.; Wang, M.; Lu, Q.B.; Wang, C.; Wu, X.; Wu, W.; Wang, Y.; Qu, W.; Han, Q.; et al. Restoring Mitochondrial Cardiolipin Homeostasis Reduces Cell Death and Promotes Recovery after Spinal Cord Injury. Cell Death Dis. 2022, 13, 1058. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Alzheimer’s Disease. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated Function of Mitochondria-Associated ER Membranes in Alzheimer Disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Yin, F. Lipid Metabolism and Alzheimer’s Disease: Clinical Evidence, Mechanistic Link and Therapeutic Promise. FEBS J. 2023, 290, 1420–1453. [Google Scholar] [CrossRef] [PubMed]
- Heverin, M.; Bogdanovic, N.; Lütjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Diczfalusy, U.; Winblad, B.; Björkhem, I. Changes in the Levels of Cerebral and Extracerebral Sterols in the Brain of Patients with Alzheimer’s Disease. J. Lipid Res. 2004, 45, 186–193. [Google Scholar] [CrossRef]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human Wild-Type Full-Length Tau Accumulation Disrupts Mitochondrial Dynamics and the Functions via Increasing Mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Tiberi, M.; Matteocci, A.; Fazio, F.; Siffeti, H.; Saracini, S.; Mercuri, N.B.; Sancesario, G. Lipidomics of Bioactive Lipids in Alzheimer’s and Parkinson’s Diseases: Where Are We? Int. J. Mol. Sci. 2022, 23, 6235. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, A.; Debbabi, M.; Bezine, M.; Karym, E.M.; Badred, A. Lipid Biomarkers in Alzheimer’s Disease Lipid Biomarkers in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 14, 303–312. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. The α-Synucleinopathies: Parkinson’s Disease, Dementia with Lewy Bodies, and Multiple System Atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein Misfolding and Aggregation: Implications in Parkinson’s Disease Pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Gilmozzi, V.; Gentile, G.; Paulina Castelo Rueda, M.; Hicks, A.A.; Pramstaller, P.P.; Zanon, A.; Lévesque, M.; Pichler, I. Interaction of Alpha-Synuclein with Lipids: Mitochondrial Cardiolipin as a Critical Player in the Pathogenesis of Parkinson’s Disease. Front. Neurosci. 2020, 14, 578993. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Kapralov, A.A.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Tyurin, V.A.; Zhao, Q.; Belikova, N.A.; Vlasova, I.I.; Maeda, A.; et al. Peroxidase Mechanism of Lipid-Dependent Cross-Linking of Synuclein with Cytochrome c. Protection against Apoptosis versus Delayed Oxidative Stress in Parkinson Disease. J. Biol. Chem. 2009, 284, 15951–15969. [Google Scholar] [CrossRef]
- Plotegher, N.; Gratton, E.; Bubacco, L. Number and Brightness Analysis of Alpha-Synuclein Oligomerization and the Associated Mitochondrial Morphology Alterations in Live Cells. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2014–2024. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Erustes, A.G.; Guarache, G.C.; Guedes, E.D.C.; Leão, A.H.F.F.; Pereira, G.J.D.S.; Smaili, S.S. α-Synuclein Interactions in Mitochondria-ER Contacts: A Possible Role in Parkinson’s Disease. Contact 2022, 5, 25152564221119347. [Google Scholar] [CrossRef]
- Avisar, H.; Guardia-Laguarta, C.; Area-Gomez, E.; Surface, M.; Chan, A.K.; Alcalay, R.N.; Lerner, B. Lipidomics Prediction of Parkinson’s Disease Severity: A Machine-Learning Analysis. J. Parkinsons. Dis. 2021, 11, 1141–1155. [Google Scholar] [CrossRef]
- Zardini Buzatto, A.; Tatlay, J.; Bajwa, B.; Mung, D.; Camicioli, R.; Dixon, R.A.; Li, L. Comprehensive Serum Lipidomics for Detecting Incipient Dementia in Parkinson’s Disease. J. Proteome Res. 2021, 20, 4053–4067. [Google Scholar] [CrossRef]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated Analyses of Microbiome and Longitudinal Metabolome Data Reveal Microbial-Host Interactions on Sulfur Metabolism in Parkinson’s Disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Robert, H.; Brown, J. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 108, 37–44. [Google Scholar] [CrossRef]
- Agrawal, I.; Lim, Y.S.; Ng, S.Y.; Ling, S.C. Deciphering Lipid Dysregulation in ALS: From Mechanisms to Translational Medicine. Transl. Neurodegener. 2022, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef]
- Cherry, S.; Jin, E.J.; Özel, M.N.; Lu, Z.; Agi, E.; Wang, D.; Jung, W.-H.; Epstein, D.; Meinertzhagen, I.A.; Chan, C.-C.; et al. Charcot-Marie-Tooth 2B Mutations in Rab7 Cause Dosage-Dependent Neurodegeneration Due to Partial Loss of Function. Elife 2013, 2, e01064. [Google Scholar] [CrossRef]
- Harris, G.; Stickland, C.A.; Lim, M.; Goldberg Oppenheimer, P. Raman Spectroscopy Spectral Fingerprints of Biomarkers of Traumatic Brain Injury. Cells 2023, 12, e01064. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-Class Cardiolipin-Protective Compound as a Therapeutic Agent to Restore Mitochondrial Bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial Protein Interaction Landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar] [CrossRef]
- Zhao, W.; Xu, Z.; Cao, J.; Fu, Q.; Wu, Y.; Zhang, X.; Long, Y.; Zhang, X.; Yang, Y.; Li, Y.; et al. Elamipretide (SS-31) Improves Mitochondrial Dysfunction, Synaptic and Memory Impairment Induced by Lipopolysaccharide in Mice. J. Neuroinflammation 2019, 16, 230. [Google Scholar] [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired Mitochondrial Biogenesis, Defective Axonal Transport of Mitochondria, Abnormal Mitochondrial Dynamics and Synaptic Degeneration in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X.; Arubala, P.; States, U.; Campus, S.W.; States, U.; States, U.; States, U.; States, U.; et al. Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease P. J. Alzheimers Dis. 2018, 62, 1549–1565. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria Targeted Peptides Protect Against. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The Cardiolipin-Binding Peptide Elamipretide Mitigates Fragmentation of Cristae Networks Following Cardiac Ischemia Reperfusion in Rats. Commun. Biol. 2020, 3, 389. [Google Scholar] [CrossRef]
- Karaa, A.; Bertini, E.; Carelli, V.; Cohen, B.H.; Enns, G.M.; Falk, M.J.; Goldstein, A.; Gorman, G.S.; Haas, R.; Hirano, M.; et al. Efficacy and Safety of Elamipretide in Individuals with Primary Mitochondrial Myopathy: The MMPOWER-3 Randomized Clinical Trial. Neurology 2023, 101, e238–e252. [Google Scholar] [CrossRef]
- Mitchell, W.; Ng, E.A.; Tamucci, J.D.; Boyd, K.J.; Sathappa, M.; Coscia, A.; Pan, M.; Han, X.; Eddy, N.A.; May, E.R.; et al. The Mitochondria-Targeted Peptide SS-31 Binds Lipid Bilayers and Modulates Surface Electrostatics as a Key Component of Its Mechanism of Action. J. Biol. Chem. 2020, 295, 7452–7469. [Google Scholar] [CrossRef]
- Gautam, M.; Genç, B.; Helmold, B.; Ahrens, A.; Kuka, J.; Makrecka-Kuka, M.; Günay, A.; Koçak, N.; Aguilar-Wickings, I.R.; Keefe, D.; et al. SBT-272 Improves TDP-43 Pathology in ALS Upper Motor Neurons by Modulating Mitochondrial Integrity, Motility, and Function. Neurobiol. Dis. 2023, 178, 106022. [Google Scholar] [CrossRef]
- Shin, G.; Hyun, S.; Kim, D.; Choi, Y.; Kim, K.H.; Kim, D.; Kwon, S.; Kim, Y.S.; Yang, S.H.; Yu, J. Cyclohexylalanine-Containing α-Helical Amphipathic Peptide Targets Cardiolipin, Rescuing Mitochondrial Dysfunction in Kidney Injury. J. Med. Chem. 2023, 67, 3385–3399. [Google Scholar] [CrossRef]
- Xun, Z.; Wipf, P.; McMurray, C.T. XJB-5-131 Is a Mild Uncoupler of Oxidative Phosphorylation. J. Huntingtons. Dis. 2022, 11, 141–151. [Google Scholar] [CrossRef]
- Stulczewski, D.; Zgorzynska, E.; Dziedzic, B.; Wieczorek-Szukala, K.; Szafraniec, K.; Walczewska, A. EPA Stronger than DHA Increases the Mitochondrial Membrane Potential and Cardiolipin Levels but Does Not Change the ATP Level in Astrocytes. Exp. Cell Res. 2023, 424, 113491. [Google Scholar] [CrossRef]
- Minamida, K.; Taira, T.; Sasaki, M.; Higuchi, O.; Meng, X.-Y.; Kamagata, Y.; Miwa, K. Extracellular Vesicles of Weizmannia Coagulans Lilac-01 Reduced Cell Death of Primary Microglia and Increased Mitochondrial Content in Dermal Fibroblasts in Vitro. Biosci. Biotechnol. Biochem. 2023, 88, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Kleinwächter, I.; Mohr, B.; Joppe, A.; Hellmann, N.; Bereau, T.; Osiewacz, H.D.; Schneider, D. CLiB—A Novel Cardiolipin-Binder Isolated via Data-Driven and in Vitro Screening. RSC Chem. Biol. 2022, 3, 941–954. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Affected | Mitochondrial Alterations | Biological Sample | Ref. |
|---|---|---|---|
| MIC10 subunit | -Cristae defects -Misdistribution of MRC | -Yeast -Hela, HepG2 cells | [29,30,31] |
| F1Fo ATP synthase | -Non-bilayer structure formation -Decrease dimerization ATP synthase in the cristae -Reduce OXPHOS | -Synthetic liposomes -Drosophila | [28,69] |
| -RCs (I, III) -RSCs (I, III, IV) | -Reduction in RCs -Misfold of RSCs | -Mouse | [70] |
| ADP/ATP carrier (AAC) | -Destabilized protein structure -OXPHOS defects. | -Yeast -Human cell lines | [39] |
| MCU, MICU1 | -Reduced mitochondrial Ca2+ uptake -Inactivation of pyruvate dehydrogenase | -BTHS patient cells and cardiac tissue | [41,42] |
| PARL | -Increase the expression of PARL leading to apoptosis | HEK293 cells | [71] |
| DRP1 | -Reduce mitochondrial fission | -Synthetic liposomes | [47] |
| p-ser579-DRP1 p-ser600-DRP1 | -Reduce DRP1 activation | -Synthetic liposomes | [72] |
| OPA1 | -Alterations mitochondrial fusion | -Synthetic liposomes | [48] |
| LC3 | -Decrease mitophagy | -Synthetic liposomes -SH-SY5Y cells | [67] |
| Cytochrome c | -Apoptosis | -HL-60 cells | [73] |
| Disease | Model | Findings | References |
|---|---|---|---|
| AD | Brain from 3xTg-AD mice | -Reduced CL species in synaptic mitochondria -Lack of detection of oxidated CL | [11] |
| SH-SY5Y-APPswedish | -Decrease in total CL content -Alterations in PG | [80] | |
| Brain from AD patients | -Slightly reduction of CL content -Decrease in FA content | [81] | |
| SH-SY5Y | -Tau protein exhibits a preference for binding to CL-rich regions of the OMM | [82] | |
| Primary microglia cultures and neuron cell lines | -CL inhibits amyloid-β secretion | [83] | |
| -Human iPSC ABCA7-KO -Brain from Abca7-KO mice | -Reduction of CL content -Decrease ATP synthesis, increase in ROS, and increase mitochondrial fusion | [84] | |
| PD | SH-SY5Y | -CL accelerates the rate of α-synuclein fibrillization, leading to hyperactive respiration | [85] |
| SNCA-mutant human pluripotent stem cells (iPSCs) and SNCA-transgenic mice | -Exposed CL to the OMM binds to and facilitates refolding of α-syn fibril. -Prolonged CL exposure in SNCA-mutants initiates recruitment of LC3 to the mitochondria and mitophagy. | [23] | |
| Freshly isolated mitochondria or liposome | -CL interacts with α-syn to favor pore formation within mitochondrial membranes | [86] | |
| N27 rat dopaminergic cell line | -CL increase α-synuclein aggregation, leading to ER stress | [87] | |
| Brain from MPTP mouse | -Inhibition of ALCAT1 prevents neurotoxicity, apoptosis, and motor deficiencies. | [88] | |
| Brain from Parkin-KO mice | -Lack of changes in CL content -CL remodeling defects with increase of short, saturated CL acyl-chains | [12,89] | |
| Brain from rat rotenone model | -Exposure to rotenone induces a loss in linoleic acid-containing CL species and an increase in CL oxidation | [16] | |
| ALS | Spinal cord from FUS mice | -Reduction in CL content | [13] |
| Cortex and spinal cord from SOD1-G86R mouse | -Reduction in CL content | [14] | |
| Serum ALS patients | -No changes in CL content -Increase in unsaturated lipids | [90] | |
| CMT2B | Fibroblasts from CMT2B patient | -Lack of measure of CL content by lipidomics -Increase in levels of unsaturated FA | [91] |
| TBI | Brain from TBI rat model | -Reduction of CL content | [77] |
| Brain from TBI patients and TBI rat models | -CL drives mitophagy | [92] | |
| SCI | Spine from SCI rat models | -Decrease in CL content and increase in CL oxidation | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuentes, J.M.; Morcillo, P. The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases. Cells 2024, 13, 609. https://doi.org/10.3390/cells13070609
Fuentes JM, Morcillo P. The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases. Cells. 2024; 13(7):609. https://doi.org/10.3390/cells13070609
Chicago/Turabian StyleFuentes, José M., and Patricia Morcillo. 2024. "The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases" Cells 13, no. 7: 609. https://doi.org/10.3390/cells13070609
APA StyleFuentes, J. M., & Morcillo, P. (2024). The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases. Cells, 13(7), 609. https://doi.org/10.3390/cells13070609