

The E3 Ubiquitin Protein Ligase LINCR Amplifies the TLR-Mediated Signals through Direct Degradation of MKP1

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmids
2.3. Antibodies and Reagents
2.4. Generation of KO Cells
2.5. Stable Cell Lines
2.6. Immunoblotting
2.7. Nuclear Extraction
2.8. Immunoprecipitation
2.9. Recombinant Protein Purification
2.10. In Vitro Ubiquitination Assay
2.11. In Vivo Ubiquitination Assay
2.12. Quantitative Real-Time PCR
2.13. Luciferase Assay
2.14. Enzyme-Linked Immunosorbent Assay (ELISA)
2.15. Statistical Analysis
3. Results
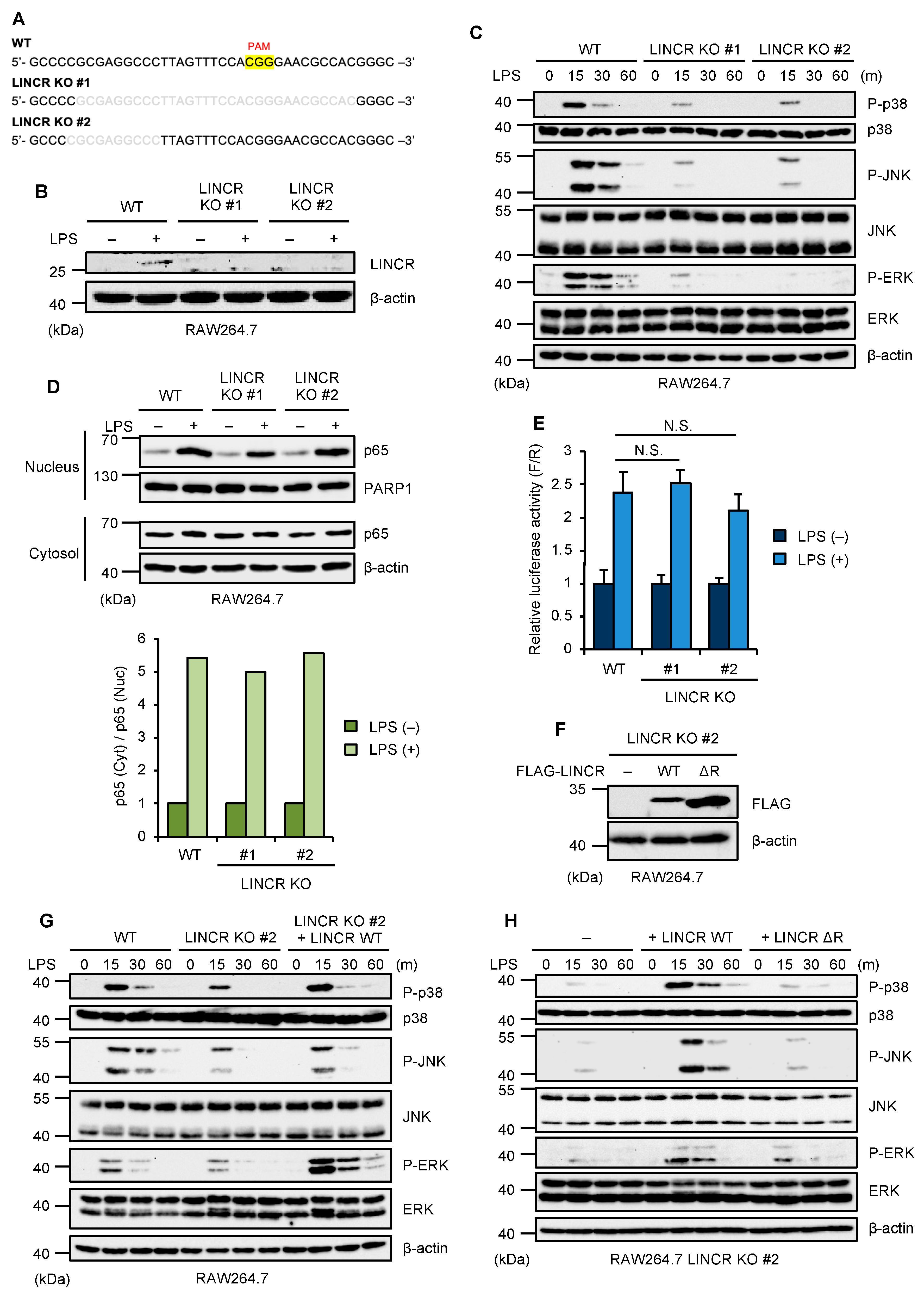
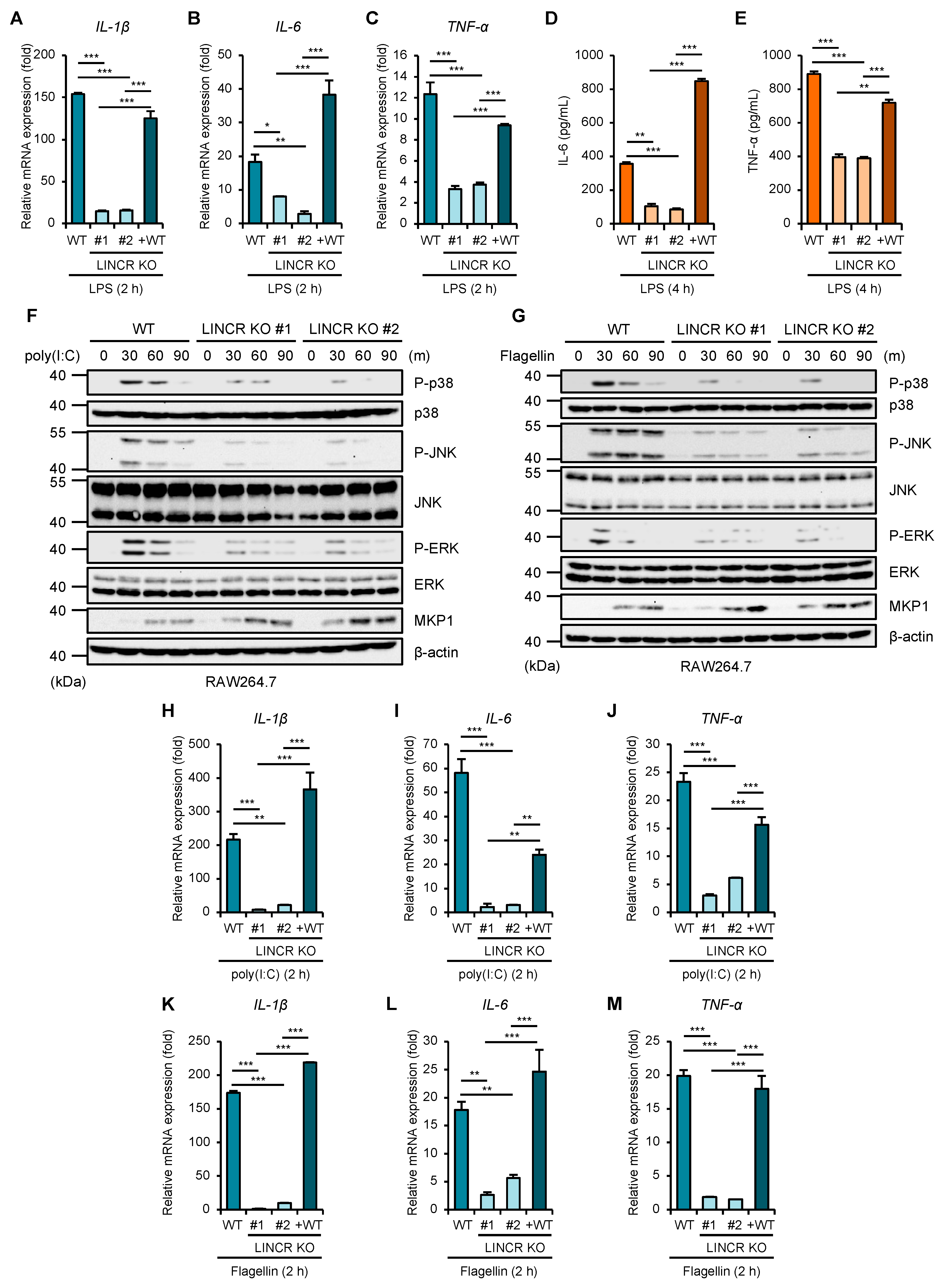
3.1. LINCR Is Required for TLR4-Mediated Activation of MAP Kinase Pathways
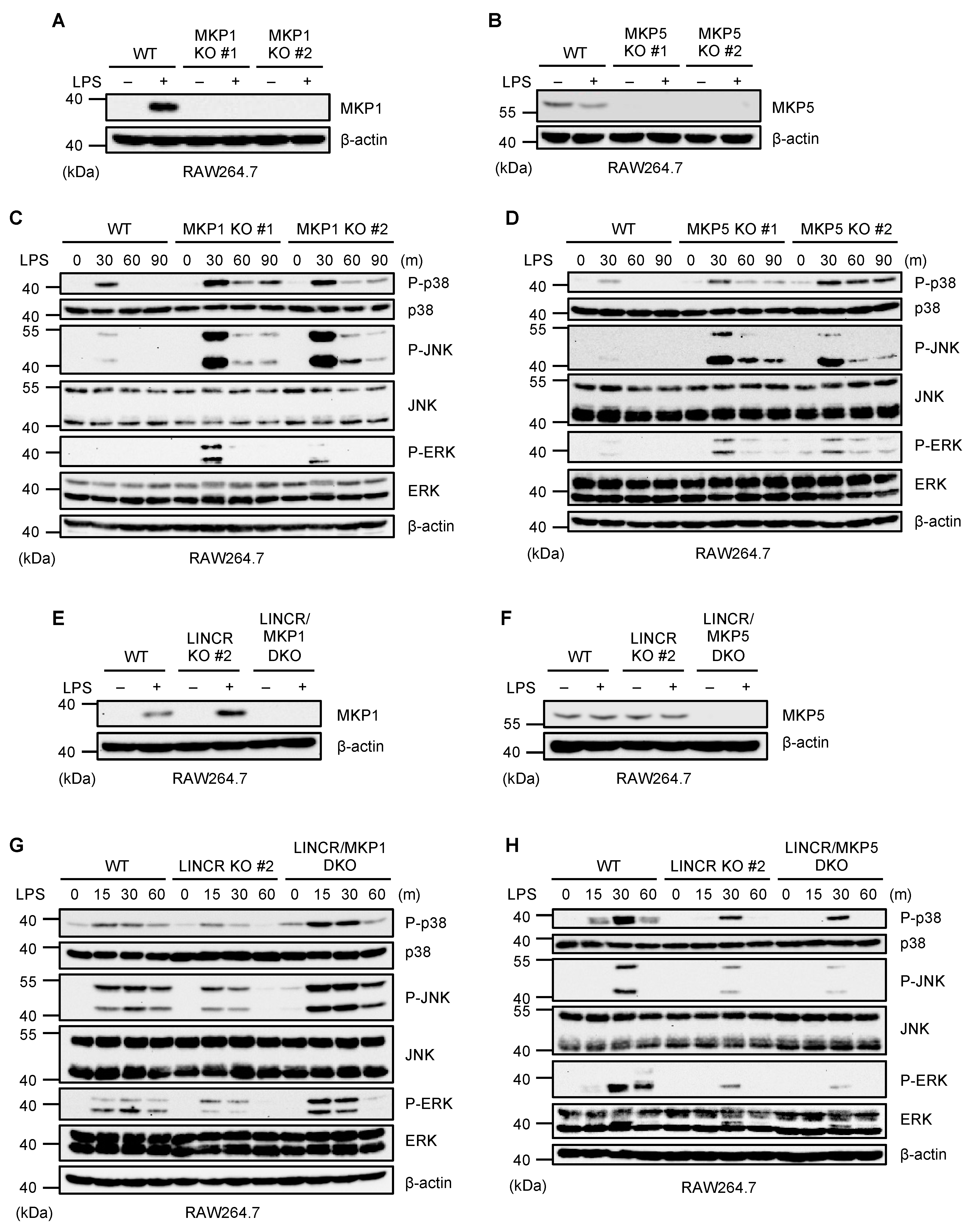
3.2. LINCR Stimulates MAP Kinase Activation by Targeting MKP1 but Not MKP5
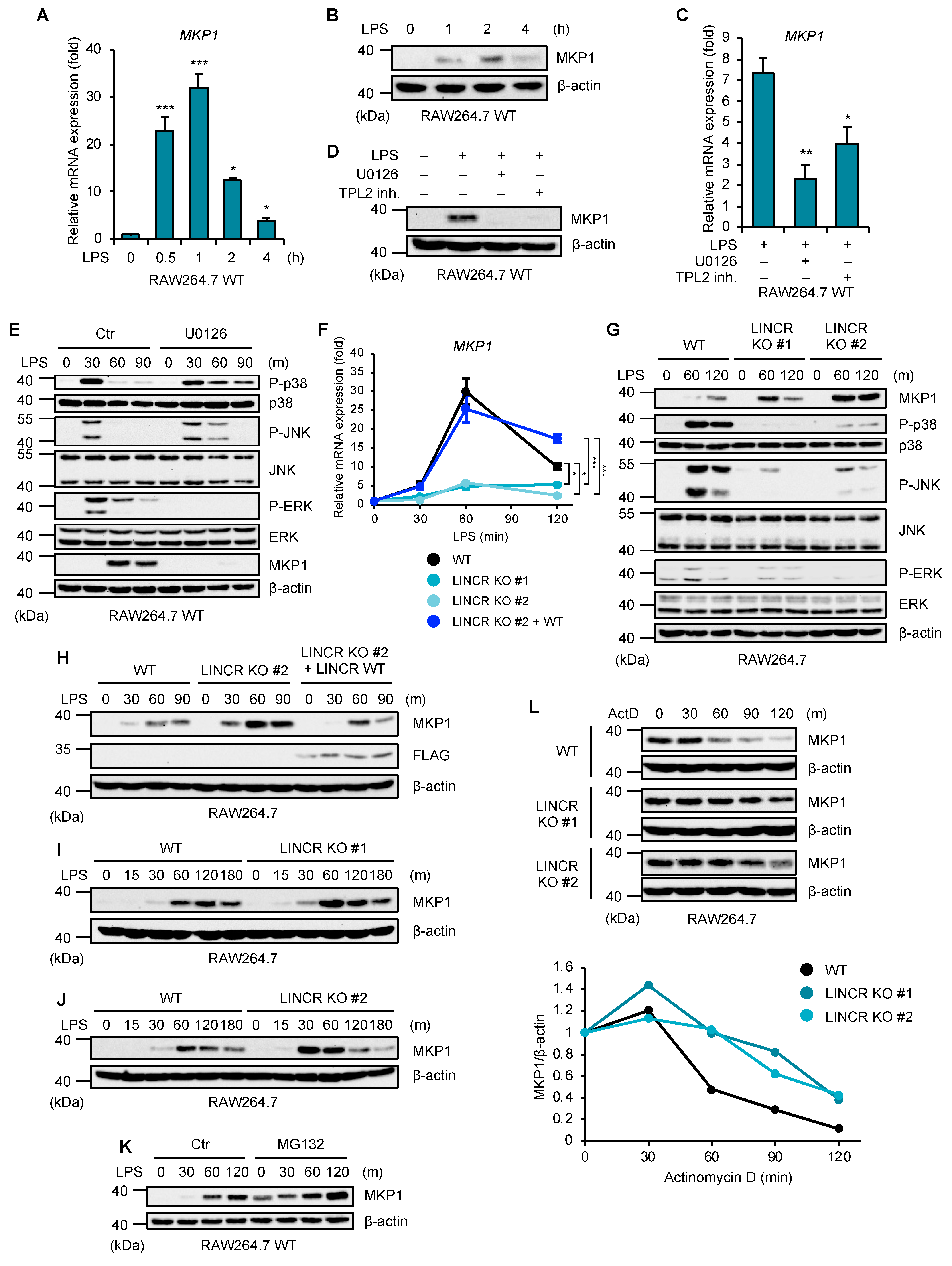
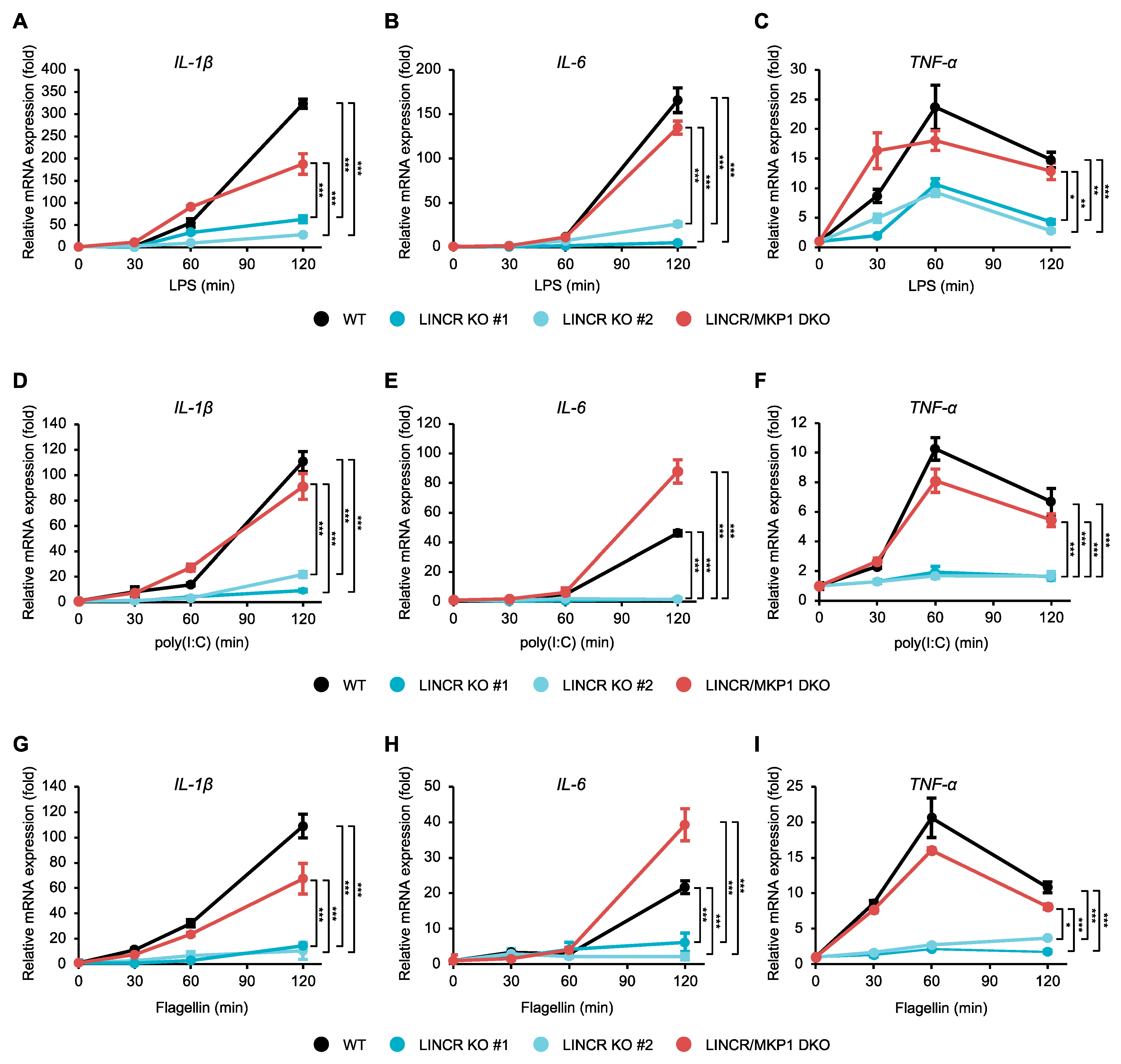
3.3. LINCR Is Involved in the Destabilization of MKP1
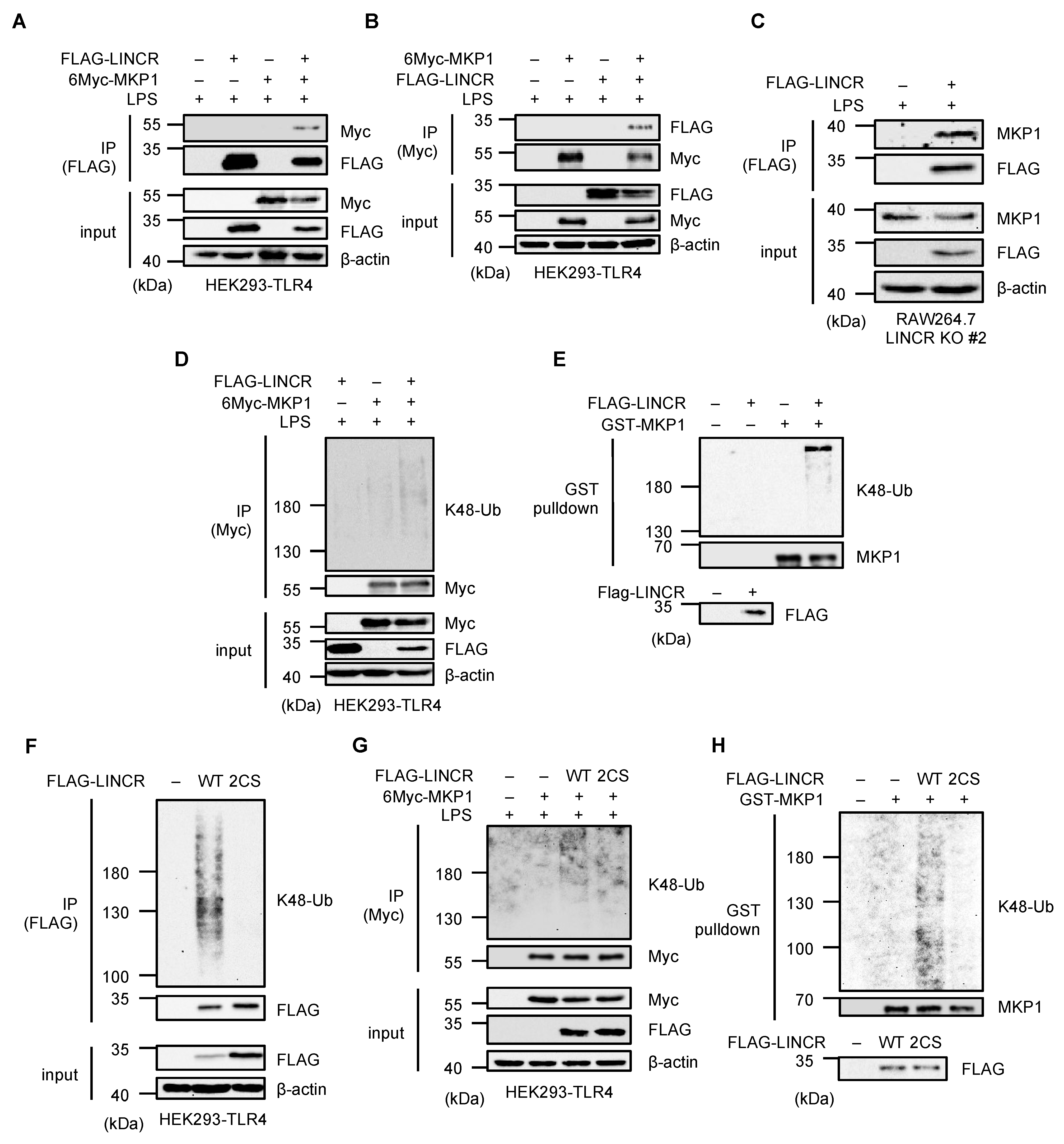
3.4. LINCR Ubiquitinates MKP1
3.5. LINCR Promotes TLR-Induced Production of a Series of Inflammatory Cytokines
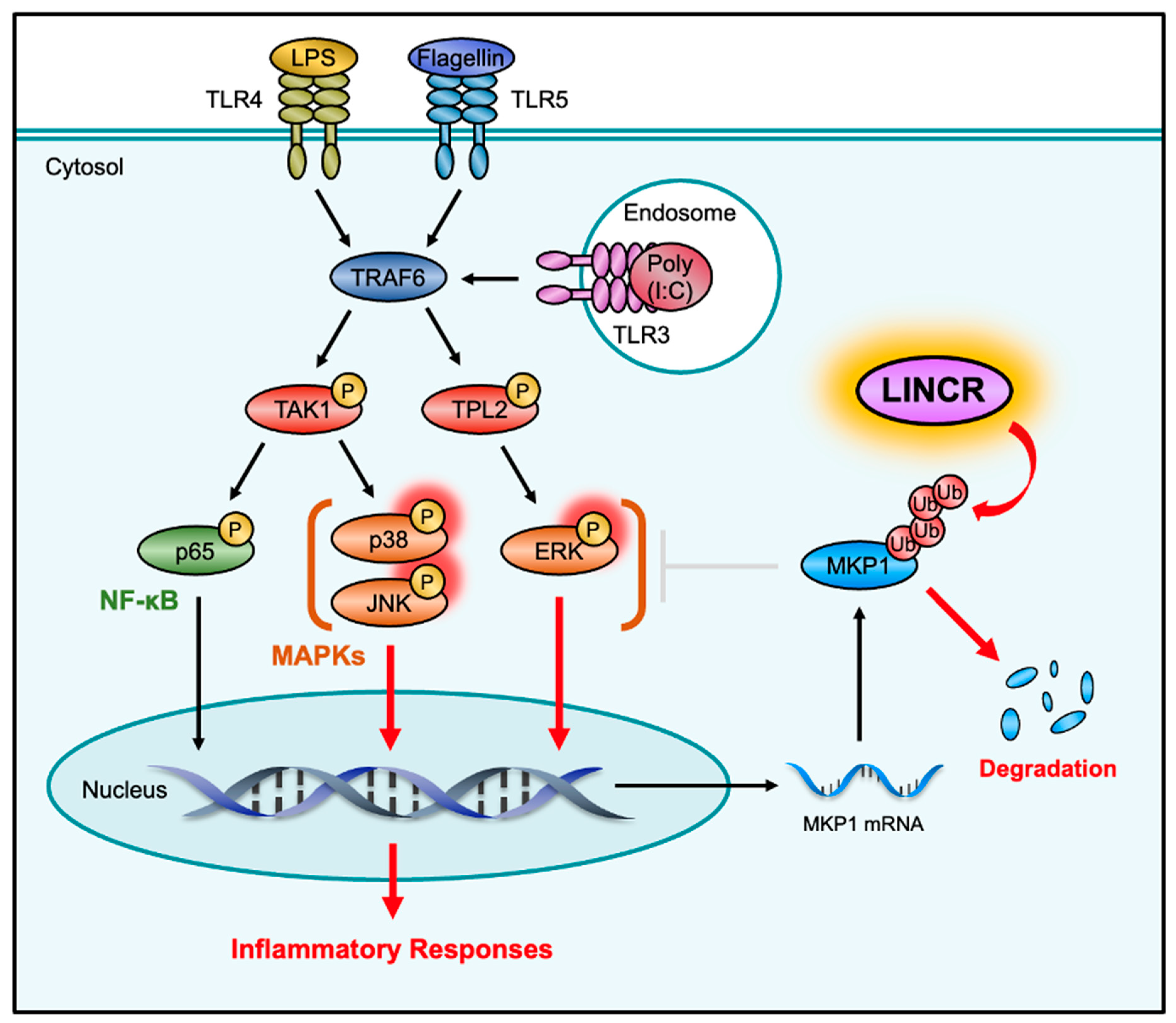
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, X.D.; Ji, T.T.; Dong, J.R.; Feng, H.; Chen, F.Q.; Chen, X.; Zhao, H.Y.; Chen, D.K.; Ma, W.T. Pathogenesis and Treatment of Cytokine Storm Induced by Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 13009. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediators Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in autoimmune diseases: A comprehensive review. Clin. Rev. Allergy Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Xu, M.; Chen, Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007, 26, 3214–3226. [Google Scholar] [CrossRef]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Saegusa, K.; Noguchi, T.; Sadamitsu, C.; Nishitoh, H.; Nagai, S.; Koyasu, S.; Matsumoto, K.; Takeda, K.; Ichijo, H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 2005, 6, 587–592. [Google Scholar] [CrossRef]
- O’Neill, L.A.J. When signaling pathways collide: Positive and negative regulation of Toll-like receptor signal transduction. Immunity 2008, 29, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. A20 restricts inflammation via ubiquitin binding. Nat. Immunol. 2020, 21, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Dikic, I. CYLD in ubiquitin signaling and tumor pathogenesis. Cell 2006, 125, 643–645. [Google Scholar] [CrossRef]
- Lang, R.; Hammer, M.; Mages, J. DUSP meet immunology: Dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol. 2006, 177, 7497–7504. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, K.L.; Camps, M.; Rommel, C.; Mackay, C.R. Targeting dual-specificity phosphatases: Manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 2007, 6, 391–403. [Google Scholar] [CrossRef]
- Crowell, S.; Wancket, L.M.; Shakibi, Y.; Xu, P.; Xue, J.; Samavati, L.; Nelin, L.D.; Liu, Y. Post-translational regulation of mitogen-activated protein kinase phosphatase (MKP)-1 and MKP-2 in macrophages following lipopolysaccharide stimulation: The role of the C termini of the phosphatases in determining their stability. J. Biol. Chem. 2014, 289, 28753–28764. [Google Scholar] [CrossRef] [PubMed]
- Brondello, J.M.; Pouyssegur, J.; McKenzie, F.R. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 1999, 286, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Chuang, S.M.; Yang, J.L. ERK1/2 achieves sustained activation by stimulating MAPK phosphatase-1 degradation via the ubiquitin-proteasome pathway. J. Biol. Chem. 2003, 278, 21534–21541. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef]
- Lin, Y.W.; Yang, J.L. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J. Biol. Chem. 2006, 281, 915–926. [Google Scholar] [CrossRef]
- Xie, P.; Guo, S.B.; Fan, Y.N.; Zhang, H.; Gu, D.F.; Li, H.H. Atrogin-1/MAFbx Enhances Simulated Ischemia/Reperfusion-induced Apoptosis in Cardiomyocytes through Degradation of MAPK Phosphatase-1 and Sustained JNK Activation. J. Biol. Chem. 2009, 284, 5488–5496. [Google Scholar] [CrossRef]
- Smith, J.B.; Nguyen, T.T.; Hughes, H.J.; Herschman, H.R.; Widney, D.P.; Bui, K.C.; Rovai, L.E. Glucocorticoid-attenuated response genes induced in the lung during endotoxemia. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L636–L647. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Nguyen, T.T.; Bui, K.C.; Demello, D.E.; Smith, J.B. A novel inflammation-induced ubiquitin E3 ligase in alveolar type II cells. Biochem. Biophys. Res. Commun. 2005, 333, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.B.; Herschman, H.R. Targeted identification of glucocorticoid-attenuated response genes: In vitro and in vivo models. Proc. Am. Thorac. Soc. 2004, 1, 275–281. [Google Scholar] [CrossRef]
- Londhe, V.A.; Tomi, T.; Nguyen, T.T.; Lopez, B.; Smith, J.B. Overexpression of LINCR in the developing mouse lung epithelium inhibits distal differentiation and induces cystic changes. Dev. Dyn. 2015, 244, 827–838. [Google Scholar] [CrossRef]
- Xu, W.; Li, H.; Dong, Z.; Cui, Z.; Zhang, N.; Meng, L.; Zhu, Y.; Liu, Y.; Li, Y.; Guo, H.; et al. Ubiquitin ligase gene neurl3 plays a role in spermatogenesis of half-smooth tongue sole (Cynoglossus semilaevis) by regulating testis protein ubiquitination. Gene 2016, 592, 215–220. [Google Scholar] [CrossRef]
- Zhao, Y.N.; Cao, X.Z.; Guo, M.Z.; Wang, X.S.; Yu, T.; Ye, L.Q.; Han, L.; Hei, L.; Tao, W.Y.; Tong, Y.M.; et al. Neuralized E3 Ubiquitin Protein Ligase 3 Is an Inducible Antiviral Effector That Inhibits Hepatitis C Virus Assembly by Targeting Viral E1 Glycoprotein. J. Virol. 2018, 92, e01123-18. [Google Scholar] [CrossRef]
- Qi, F.; Zhang, X.; Wang, L.K.; Ren, C.X.; Zhao, X.Y.; Luo, J.Y.; Lu, D. E3 ubiquitin ligase NEURL3 promotes innate antiviral response through catalyzing K63-linked ubiquitination of IRF7. FASEB J. 2022, 36, e22409. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Suzuki, M.; Mutoh, N.; Hirata, Y.; Tsuchida, M.; Miyagawa, S.; Hwang, G.; Aoki, J.; Matsuzawa, A. Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Noguchi, T.; Sekiguchi, Y.; Shimada, T.; Suzuki, W.; Yokosawa, T.; Itoh, T.; Yamada, M.; Suzuki, M.; Kurokawa, R.; Hirata, Y.; et al. LLPS of SQSTM1/p62 and NBR1 as outcomes of lysosomal stress response limits cancer cell metastasis. Proc. Natl. Acad. Sci. USA 2023, 120, e2311282120. [Google Scholar] [CrossRef]
- Yamada, Y.; Noguchi, T.; Suzuki, M.; Yamada, M.; Hirata, Y.; Matsuzawa, A. Reactive sulfur species disaggregate the SQSTM1/p62-based aggresome-like induced structures via the HSP70 induction and prevent parthanatos. J. Biol. Chem. 2023, 299, 104710. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Sekiguchi, Y.; Kudoh, Y.; Naganuma, R.; Kagi, T.; Nishidate, A.; Maeda, K.; Ishii, C.; Toyama, T.; Hirata, Y.; et al. Gefitinib initiates sterile inflammation by promoting IL-1beta and HMGB1 release via two distinct mechanisms. Cell Death Dis. 2021, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Asai, Y.; Kagi, T.; Noguchi, T.; Yamada, M.; Hirata, Y.; Matsuzawa, A. TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms. Int. J. Mol. Sci. 2020, 21, 9497. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.D.; Ceci, J.D.; Tsatsanis, C.; Kontoyiannis, D.; Stamatakis, K.; Lin, J.H.; Patriotis, C.; Jenkins, N.A.; Copeland, N.G.; Kollias, G.; et al. TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 2000, 103, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Shepherd, E.G.; Manson, M.E.; Nelin, L.D.; Sorokin, A.; Liu, Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: Attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J. Biol. Chem. 2005, 280, 8101–8108. [Google Scholar] [CrossRef] [PubMed]
- Mues, N.; Chu, H.W. Out-Smarting the Host: Bacteria Maneuvering the Immune Response to Favor Their Survival. Front. Immunol. 2020, 11, 819. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef]
- Ohtake, F. Branched ubiquitin code: From basic biology to targeted protein degradation. J. Biochem. 2022, 171, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Matta, R.; Barnard, J.A.; Wancket, L.M.; Yan, J.; Xue, J.; Grieves, J.; Frazier, W.J.; Nelin, L.; Cato, A.C.; Liu, Y. Knockout of Mkp-1 exacerbates colitis in Il-10-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1322–G1335. [Google Scholar] [CrossRef] [PubMed]
- Kjellerup, R.B.; Johansen, C.; Kragballe, K.; Iversen, L. The expression of dual-specificity phosphatase 1 mRNA is downregulated in lesional psoriatic skin. Br. J. Dermatol. 2013, 168, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Hoppstadter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef]
- Wang, X.; Nelin, L.D.; Kuhlman, J.R.; Meng, X.; Welty, S.E.; Liu, Y. The role of MAP kinase phosphatase-1 in the protective mechanism of dexamethasone against endotoxemia. Life Sci. 2008, 83, 671–680. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokosawa, T.; Miyagawa, S.; Suzuki, W.; Nada, Y.; Hirata, Y.; Noguchi, T.; Matsuzawa, A. The E3 Ubiquitin Protein Ligase LINCR Amplifies the TLR-Mediated Signals through Direct Degradation of MKP1. Cells 2024, 13, 687. https://doi.org/10.3390/cells13080687
Yokosawa T, Miyagawa S, Suzuki W, Nada Y, Hirata Y, Noguchi T, Matsuzawa A. The E3 Ubiquitin Protein Ligase LINCR Amplifies the TLR-Mediated Signals through Direct Degradation of MKP1. Cells. 2024; 13(8):687. https://doi.org/10.3390/cells13080687
Chicago/Turabian StyleYokosawa, Takumi, Sayoko Miyagawa, Wakana Suzuki, Yuki Nada, Yusuke Hirata, Takuya Noguchi, and Atsushi Matsuzawa. 2024. "The E3 Ubiquitin Protein Ligase LINCR Amplifies the TLR-Mediated Signals through Direct Degradation of MKP1" Cells 13, no. 8: 687. https://doi.org/10.3390/cells13080687
APA StyleYokosawa, T., Miyagawa, S., Suzuki, W., Nada, Y., Hirata, Y., Noguchi, T., & Matsuzawa, A. (2024). The E3 Ubiquitin Protein Ligase LINCR Amplifies the TLR-Mediated Signals through Direct Degradation of MKP1. Cells, 13(8), 687. https://doi.org/10.3390/cells13080687