
Targeting Interactions between Fibroblasts and Macrophages to Treat Cardiac Fibrosis

Abstract
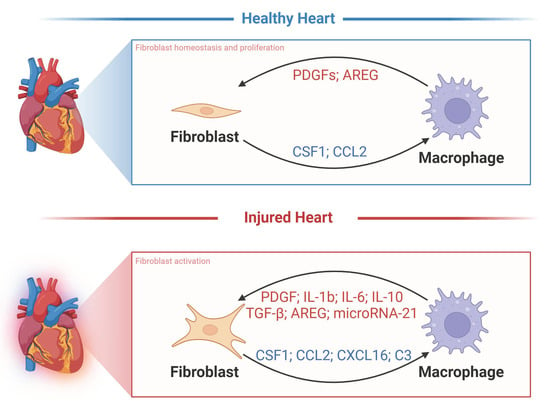
:
1. Introduction
2. Cellular Composition of the Heart
2.1. Fibroblasts in Cardiac Tissue
2.2. Macrophages in Cardiac Tissue
3. Interactions between Cardiac Macrophages and Fibroblasts
3.1. TGFβ
3.2. IL-4 and IL-6
3.3. IL-17A
3.4. MMP-2, MMP-9, and MMP-12
3.5. CX3CR 1
3.6. microRNA-21
3.7. microRNA-155
3.8. TLR2
4. Fibroblast–Macrophage Interactions for Novel Fibrosis Treatments
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Verheule, S.; Schotten, U. Electrophysiological Consequences of Cardiac Fibrosis. Cells 2021, 10, 3220. [Google Scholar] [CrossRef]
- Weber, K.T.; Pick, R.; Jalil, J.E.; Janicki, J.S.; Carroll, E.P. Patterns of myocardial fibrosis. J. Mol. Cell. Cardiol. 1989, 21 (Suppl. S5), 121–131. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Aguiar, C.M.; Gawdat, K.; Legere, S.; Marshall, J.; Hassan, A.; Kienesberger, P.C.; Pulinilkunnil, T.; Castonguay, M.; Brunt, K.R.; Legare, J.F. Fibrosis independent atrial fibrillation in older patients is driven by substrate leukocyte infiltration: Diagnostic and prognostic implications to patients undergoing cardiac surgery. J. Transl. Med. 2019, 17, 413. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Mollmann, H.; Nef, H.M.; Kostin, S.; von Kalle, C.; Pilz, I.; Weber, M.; Schaper, J.; Hamm, C.W.; Elsasser, A. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc. Res. 2006, 71, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G.; et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289. [Google Scholar] [CrossRef]
- Haudek, S.B.; Cheng, J.; Du, J.; Wang, Y.; Hermosillo-Rodriguez, J.; Trial, J.; Taffet, G.E.; Entman, M.L. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2010, 49, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Trial, J.; Entman, M.L.; Cieslik, K.A. Mesenchymal stem cell-derived inflammatory fibroblasts mediate interstitial fibrosis in the aging heart. J. Mol. Cell. Cardiol. 2016, 91, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Models Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef]
- Haider, N.; Bosca, L.; Zandbergen, H.R.; Kovacic, J.C.; Narula, N.; Gonzalez-Ramos, S.; Fernandez-Velasco, M.; Agrawal, S.; Paz-Garcia, M.; Gupta, S.; et al. Transition of Macrophages to Fibroblast-Like Cells in Healing Myocardial Infarction. J. Am. Coll. Cardiol. 2019, 74, 3124–3135. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.L.; McCrea, J.C.; Hedrick, H.L. Age-associated changes in cardiac matrix and integrins. Mech. Ageing Dev. 2001, 122, 1739–1756. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.J.; Dangerfield, A.L.; Rattan, S.G.; Bathe, K.L.; Cunnington, R.H.; Raizman, J.E.; Bedosky, K.M.; Freed, D.H.; Kardami, E.; Dixon, I.M. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev. Dyn. 2010, 239, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Yue, Y.; Yang, X.; Feng, K.; Wang, L.; Hou, J.; Mei, B.; Qin, H.; Liang, M.; Chen, G.; Wu, Z. M2b macrophages reduce early reperfusion injury after myocardial ischemia in mice: A predominant role of inhibiting apoptosis via A20. Int. J. Cardiol. 2017, 245, 228–235, Corrigendum in Int. J. Cardiol. 2019, 278, 311. [Google Scholar] [CrossRef]
- Mahmoud, A.I.; Porrello, E.R. Turning back the cardiac regenerative clock: Lessons from the neonate. Trends Cardiovasc. Med. 2012, 22, 128–133. [Google Scholar] [CrossRef]
- Wei, K.H.; Lin, I.T.; Chowdhury, K.; Lim, K.L.; Liu, K.T.; Ko, T.M.; Chang, Y.M.; Yang, K.C.; Lai, S.B. Comparative single-cell profiling reveals distinct cardiac resident macrophages essential for zebrafish heart regeneration. eLife 2023, 12, e84679. [Google Scholar] [CrossRef]
- Drenckhahn, J.D.; Schwarz, Q.P.; Gray, S.; Laskowski, A.; Kiriazis, H.; Ming, Z.; Harvey, R.P.; Du, X.J.; Thorburn, D.R.; Cox, T.C. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev. Cell 2008, 15, 521–533. [Google Scholar] [CrossRef]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Gessain, G.; Bleriot, C.; Ginhoux, F. Non-genetic Heterogeneity of Macrophages in Diseases—A Medical Perspective. Front. Cell Dev. Biol. 2020, 8, 613116. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.W.; Schroeder, A.V.; Smith-Berdan, S.; Forsberg, E.C. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 2011, 9, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Boiers, C.; Carrelha, J.; Lutteropp, M.; Luc, S.; Green, J.C.; Azzoni, E.; Woll, P.S.; Mead, A.J.; Hultquist, A.; Swiers, G.; et al. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell 2013, 13, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, I.M.; Samokhvalova, N.I.; Nishikawa, S. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 2007, 446, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages Are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef]
- Jia, D.; Chen, S.; Bai, P.; Luo, C.; Liu, J.; Sun, A.; Ge, J. Cardiac Resident Macrophage-Derived Legumain Improves Cardiac Repair by Promoting Clearance and Degradation of Apoptotic Cardiomyocytes after Myocardial Infarction. Circulation 2022, 145, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Revelo, X.S.; Parthiban, P.; Chen, C.; Barrow, F.; Fredrickson, G.; Wang, H.; Yucel, D.; Herman, A.; van Berlo, J.H. Cardiac Resident Macrophages Prevent Fibrosis and Stimulate Angiogenesis. Circ. Res. 2021, 129, 1086–1101. [Google Scholar] [CrossRef]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Chitu, V.; Stanley, E.R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006, 18, 39–48. [Google Scholar] [CrossRef]
- Meziani, L.; Mondini, M.; Petit, B.; Boissonnas, A.; Thomas de Montpreville, V.; Mercier, O.; Vozenin, M.C.; Deutsch, E. CSF1R inhibition prevents radiation pulmonary fibrosis by depletion of interstitial macrophages. Eur. Respir. J. 2018, 51, 1702120. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Fu, W.; Turley, S.J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 2021, 54, 903–915. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- McKleroy, W.; Lee, T.H.; Atabai, K. Always cleave up your mess: Targeting collagen degradation to treat tissue fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L709–L721. [Google Scholar] [CrossRef]
- Atabai, K.; Jame, S.; Azhar, N.; Kuo, A.; Lam, M.; McKleroy, W.; Dehart, G.; Rahman, S.; Xia, D.D.; Melton, A.C.; et al. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Investig. 2009, 119, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Vivar, R.; Boza, P.; Munoz, C.; Bolivar, S.; Anfossi, R.; Osorio, J.M.; Olivares-Silva, F.; Garcia, L.; Diaz-Araya, G. Cardiac fibroblast cytokine profiles induced by proinflammatory or profibrotic stimuli promote monocyte recruitment and modulate macrophage M1/M2 balance in vitro. J. Mol. Cell. Cardiol. 2016, 101, 69–80. [Google Scholar] [CrossRef]
- Ceccato, T.L.; Starbuck, R.B.; Hall, J.K.; Walker, C.J.; Brown, T.E.; Killgore, J.P.; Anseth, K.S.; Leinwand, L.A. Defining the Cardiac Fibroblast Secretome in a Fibrotic Microenvironment. J. Am. Heart Assoc. 2020, 9, e017025. [Google Scholar] [CrossRef]
- Vi, L.; Baht, G.S.; Soderblom, E.J.; Whetstone, H.; Wei, Q.; Furman, B.; Puviindran, V.; Nadesan, P.; Foster, M.; Poon, R.; et al. Macrophage cells secrete factors including LRP1 that orchestrate the rejuvenation of bone repair in mice. Nat. Commun. 2018, 9, 5191. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-β1. Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef]
- Meng, Q.; Bhandary, B.; Bhuiyan, M.S.; James, J.; Osinska, H.; Valiente-Alandi, I.; Shay-Winkler, K.; Gulick, J.; Molkentin, J.D.; Blaxall, B.C.; et al. Myofibroblast-Specific TGFβ Receptor II Signaling in the Fibrotic Response to Cardiac Myosin Binding Protein C-Induced Cardiomyopathy. Circ. Res. 2018, 123, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Munoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Frangogiannis, N. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Fichtner-Feigl, S.; Strober, W.; Kawakami, K.; Puri, R.K.; Kitani, A. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat. Med. 2006, 12, 99–106. [Google Scholar] [CrossRef]
- Brunner, S.M.; Schiechl, G.; Kesselring, R.; Martin, M.; Balam, S.; Schlitt, H.J.; Geissler, E.K.; Fichtner-Feigl, S. IL-13 signaling via IL-13Rα2 triggers TGF-β1-dependent allograft fibrosis. Transplant. Res. 2013, 2, 16. [Google Scholar] [CrossRef]
- Wu, L.; Ong, S.; Talor, M.V.; Barin, J.G.; Baldeviano, G.C.; Kass, D.A.; Bedja, D.; Zhang, H.; Sheikh, A.; Margolick, J.B.; et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J. Exp. Med. 2014, 211, 1449–1464. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef]
- de Couto, G.; Liu, W.; Tseliou, E.; Sun, B.; Makkar, N.; Kanazawa, H.; Arditi, M.; Marban, E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J. Clin. Investig. 2015, 125, 3147–3162. [Google Scholar] [CrossRef] [PubMed]
- Tseliou, E.; de Couto, G.; Terrovitis, J.; Sun, B.; Weixin, L.; Marban, L.; Marban, E. Angiogenesis, cardiomyocyte proliferation and anti-fibrotic effects underlie structural preservation post-infarction by intramyocardially-injected cardiospheres. PLoS ONE 2014, 9, e88590. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Tseliou, E.; Malliaras, K.; Yee, K.; Dawkins, J.F.; De Couto, G.; Smith, R.R.; Kreke, M.; Seinfeld, J.; Middleton, R.C.; et al. Cellular postconditioning: Allogeneic cardiosphere-derived cells reduce infarct size and attenuate microvascular obstruction when administered after reperfusion in pigs with acute myocardial infarction. Circ. Heart Fail. 2015, 8, 322–332. [Google Scholar] [CrossRef]
- Kubota, A.; Suto, A.; Suzuki, K.; Kobayashi, Y.; Nakajima, H. Matrix metalloproteinase-12 produced by Ly6Clow macrophages prolongs the survival after myocardial infarction by preventing neutrophil influx. J. Mol. Cell. Cardiol. 2019, 131, 41–52. [Google Scholar] [CrossRef]
- Falkenham, A.; Myers, T.; Wong, C.; Legare, J.F. Implications for the role of macrophages in a model of myocardial fibrosis: CCR2−/− mice exhibit an M2 phenotypic shift in resident cardiac macrophages. Cardiovasc. Pathol. 2016, 25, 390–398. [Google Scholar] [CrossRef]
- Kumar, S.; Wang, G.; Zheng, N.; Cheng, W.; Ouyang, K.; Lin, H.; Liao, Y.; Liu, J. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension 2019, 73, 1058–1070. [Google Scholar] [CrossRef]
- Tao, Z.Y.; Cavasin, M.A.; Yang, F.; Liu, Y.H.; Yang, X.P. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004, 74, 1561–1572. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Liu, L.; Xi, A.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Y.L.; Zhang, C.C.; Cui, W.; Wang, X.; Xia, Y.; Du, J.; Li, H.H. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc. Res. 2014, 101, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A. The Many Alternative Faces of Macrophage Activation. Front. Immunol. 2015, 6, 370. [Google Scholar] [CrossRef]
- Mosser, D.M. The many faces of macrophage activation. J. Leucoc. Biol. 2003, 73, 209–212. [Google Scholar] [CrossRef]
- Goldmann, O.; von Kockritz-Blickwede, M.; Holtje, C.; Chhatwal, G.S.; Geffers, R.; Medina, E. Transcriptome analysis of murine macrophages in response to infection with Streptococcus pyogenes reveals an unusual activation program. Infect. Immun. 2007, 75, 4148–4157. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.F.; Mosser, D.M. A novel phenotype for an activated macrophage: The type 2 activated macrophage. J. Leukoc. Biol. 2002, 72, 101–106. [Google Scholar] [CrossRef]
- Song, J.; Frieler, R.A.; Whitesall, S.E.; Chung, Y.; Vigil, T.M.; Muir, L.A.; Ma, J.; Brombacher, F.; Goonewardena, S.N.; Lumeng, C.N.; et al. Myeloid interleukin-4 receptor α is essential in postmyocardial infarction healing by regulating inflammation and fibrotic remodeling. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H323–H337. [Google Scholar] [CrossRef]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef]
- Hirota, H.; Yoshida, K.; Kishimoto, T.; Taga, T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc. Natl. Acad. Sci. USA 1995, 92, 4862–4866. [Google Scholar] [CrossRef]
- Huo, S.; Shi, W.; Ma, H.; Yan, D.; Luo, P.; Guo, J.; Li, C.; Lin, J.; Zhang, C.; Li, S.; et al. Alleviation of Inflammation and Oxidative Stress in Pressure Overload-Induced Cardiac Remodeling and Heart Failure via IL-6/STAT3 Inhibition by Raloxifene. Oxid. Med. Cell Longev. 2021, 2021, 6699054. [Google Scholar] [CrossRef]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Schmidl, C.; Renner, K.; Peter, K.; Eder, R.; Lassmann, T.; Balwierz, P.J.; Itoh, M.; Nagao-Sato, S.; Kawaji, H.; Carninci, P.; et al. Transcription and enhancer profiling in human monocyte subsets. Blood 2014, 123, e90–e99. [Google Scholar] [CrossRef] [PubMed]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012, 92, 635–688. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, G.; Bracamonte-Baran, W.; Choi, H.S.; Diny, N.L.; Sung, J.; Hughes, D.; Won, T.; Wood, M.K.; Talor, M.V.; et al. The Cardiac Microenvironment Instructs Divergent Monocyte Fates and Functions in Myocarditis. Cell Rep. 2019, 28, 172–189.e7. [Google Scholar] [CrossRef] [PubMed]
- Aimes, R.T.; Quigley, J.P. Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J. Biol. Chem. 1995, 270, 5872–5876. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [CrossRef]
- Li, Y.; Li, Q.; Fan, G.C. Macrophage Efferocytosis in Cardiac Pathophysiology and Repair. Shock 2021, 55, 177–188. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Jeyalan, V.; Austin, D.; Loh, S.X.; Wangsaputra, V.K.; Spyridopoulos, I. Fractalkine/CX3CR1 in Dilated Cardiomyopathy: A Potential Future Target for Immunomodulatory Therapy? Cells 2023, 12, 2377. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jurgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A.; et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef]
- Philippen, L.E.; Dirkx, E.; Wit, J.B.; Burggraaf, K.; de Windt, L.J.; da Costa Martins, P.A. Antisense MicroRNA Therapeutics in Cardiovascular Disease: Quo Vadis? Mol. Ther. 2015, 23, 1810–1818. [Google Scholar] [CrossRef]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef]
- Hinkel, R.; Ramanujam, D.; Kaczmarek, V.; Howe, A.; Klett, K.; Beck, C.; Dueck, A.; Thum, T.; Laugwitz, K.L.; Maegdefessel, L.; et al. AntimiR-21 Prevents Myocardial Dysfunction in a Pig Model of Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2020, 75, 1788–1800. [Google Scholar] [CrossRef]
- Ramanujam, D.; Sassi, Y.; Laggerbauer, B.; Engelhardt, S. Viral Vector-Based Targeting of miR-21 in Cardiac Nonmyocyte Cells Reduces Pathologic Remodeling of the Heart. Mol. Ther. 2016, 24, 1939–1948. [Google Scholar] [CrossRef]
- Ramanujam, D.; Schon, A.P.; Beck, C.; Vaccarello, P.; Felician, G.; Dueck, A.; Esfandyari, D.; Meister, G.; Meitinger, T.; Schulz, C.; et al. MicroRNA-21-Dependent Macrophage-to-Fibroblast Signaling Determines the Cardiac Response to Pressure Overload. Circulation 2021, 143, 1513–1525. [Google Scholar] [CrossRef]
- Malik, J.A.; Kaur, G.; Agrewala, J.N. Revolutionizing medicine with toll-like receptors: A path to strengthening cellular immunity. Int. J. Biol. Macromol. 2023, 253, 127252. [Google Scholar] [CrossRef] [PubMed]
- De Blasio, M.J.; Ohlstein, E.H.; Ritchie, R.H. Therapeutic targets of fibrosis: Translational advances and current challenges. Br. J. Pharmacol. 2023, 180, 2839–2845. [Google Scholar] [CrossRef] [PubMed]
- Pfau, D.; Thorn, S.L.; Zhang, J.; Mikush, N.; Renaud, J.M.; Klein, R.; deKemp, R.A.; Wu, X.; Hu, X.; Sinusas, A.J.; et al. Angiotensin Receptor Neprilysin Inhibitor Attenuates Myocardial Remodeling and Improves Infarct Perfusion in Experimental Heart Failure. Sci. Rep. 2019, 9, 5791. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.P.; Prescott, M.F.; Maisel, A.S.; Butler, J.; Pina, I.L.; Felker, G.M.; Ward, J.H.; Williamson, K.M.; Camacho, A.; Kandanelly, R.R.; et al. Association between Angiotensin Receptor-Neprilysin Inhibition, Cardiovascular Biomarkers, and Cardiac Remodeling in Heart Failure with Reduced Ejection Fraction. Circ. Heart Fail. 2021, 14, e008410. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Mason, T.; Coelho-Filho, O.R.; Verma, S.; Chowdhury, B.; Zuo, F.; Quan, A.; Thorpe, K.E.; Bonneau, C.; Teoh, H.; Gilbert, R.E.; et al. Empagliflozin Reduces Myocardial Extracellular Volume in Patients with Type 2 Diabetes and Coronary Artery Disease. JACC Cardiovasc. Imaging 2021, 14, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Ravassa, S.; Trippel, T.; Bach, D.; Bachran, D.; Gonzalez, A.; Lopez, B.; Wachter, R.; Hasenfuss, G.; Delles, C.; Dominiczak, A.F.; et al. Biomarker-based phenotyping of myocardial fibrosis identifies patients with heart failure with preserved ejection fraction resistant to the beneficial effects of spironolactone: Results from the Aldo-DHF trial. Eur. J. Heart Fail. 2018, 20, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhao, S.H.; Lu, M.J. Update on the role of CMR in the evaluation of heart failure: Interpretation of the “2022 AHA/ACC/HFSA Guideline for the management of heart failure”. Zhonghua Xin Xue Guan Bing Za Zhi 2023, 51, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Rudebusch, J.; Benkner, A.; Nath, N.; Fleuch, L.; Kaderali, L.; Grube, K.; Klingel, K.; Eckstein, G.; Meitinger, T.; Fielitz, J.; et al. Stimulation of soluble guanylyl cyclase (sGC) by riociguat attenuates heart failure and pathological cardiac remodelling. Br. J. Pharmacol. 2022, 179, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hang, W.; Shu, H.; Zhou, N. Pirfenidone alleviates cardiac fibrosis induced by pressure overload via inhibiting TGF-β1/Smad3 signalling pathway. J. Cell Mol. Med. 2022, 26, 4548–4555. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, M.; Ito, H.; Onuki, T.; Miyoshi, F.; Watanabe, N.; Asano, T.; Tanno, K.; Kobayashi, Y. Candesartan decreases type III procollagen-N-peptide levels and inflammatory marker levels and maintains sinus rhythm in patients with atrial fibrillation. J. Cardiovasc. Pharmacol. 2010, 55, 511–517. [Google Scholar] [CrossRef]
- Kosmala, W.; Przewlocka-Kosmala, M.; Szczepanik-Osadnik, H.; Mysiak, A.; O’Moore-Sullivan, T.; Marwick, T.H. A randomized study of the beneficial effects of aldosterone antagonism on LV function, structure, and fibrosis markers in metabolic syndrome. JACC Cardiovasc. Imaging 2011, 4, 1239–1249. [Google Scholar] [CrossRef]
- Deswal, A.; Richardson, P.; Bozkurt, B.; Mann, D.L. Results of the Randomized Aldosterone Antagonism in Heart Failure with Preserved Ejection Fraction trial (RAAM-PEF). J. Card. Fail. 2011, 17, 634–642. [Google Scholar] [CrossRef]
- Shimada, Y.J.; Passeri, J.J.; Baggish, A.L.; O’Callaghan, C.; Lowry, P.A.; Yannekis, G.; Abbara, S.; Ghoshhajra, B.B.; Rothman, R.D.; Ho, C.Y.; et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013, 1, 480–487. [Google Scholar] [CrossRef]
- Kosmala, W.; Przewlocka-Kosmala, M.; Szczepanik-Osadnik, H.; Mysiak, A.; Marwick, T.H. Fibrosis and cardiac function in obesity: A randomised controlled trial of aldosterone blockade. Heart 2013, 99, 320–326. [Google Scholar] [CrossRef]
- Fang, L.; Murphy, A.J.; Dart, A.M. A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease. Front. Pharmacol. 2017, 8, 186. [Google Scholar] [CrossRef]
- Simonetti, J.; Sgalla, G.; Richeldi, L. An up-to-date review of approved and emerging antibody therapies for idiopathic pulmonary fibrosis. Expert Opin. Biol. Ther. 2023, 23, 1239–1244. [Google Scholar] [CrossRef]
- Zhao, C.; Yin, Y.; Zhu, C.; Zhu, M.; Ji, T.; Li, Z.; Cai, J. Drug therapies for treatment of idiopathic pulmonary fibrosis: A systematic review, Bayesian network meta-analysis, and cost-effectiveness analysis. EClinicalMedicine 2023, 61, 102071. [Google Scholar] [CrossRef]
- Lewis, G.A.; Dodd, S.; Clayton, D.; Bedson, E.; Eccleson, H.; Schelbert, E.B.; Naish, J.H.; Jimenez, B.D.; Williams, S.G.; Cunnington, C.; et al. Pirfenidone in heart failure with preserved ejection fraction: A randomized phase 2 trial. Nat. Med. 2021, 27, 1477–1482. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, H.; Huang, X.; Wang, S.; Ouyang, X.; Wang, Y.; Cao, Q.; Yang, L.; Tao, Y.; Lai, H. Pirfenidone attenuates cardiac hypertrophy against isoproterenol by inhibiting activation of the janus tyrosine kinase-2/signal transducer and activator of transcription 3 (JAK-2/STAT3) signaling pathway. Bioengineered 2022, 13, 12772–12782. [Google Scholar] [CrossRef] [PubMed]
- Soltani, F.; Lewis, G.A.; Rosala-Hallas, A.; Dodd, S.; Schelbert, E.B.; Williams, S.G.; Cunnington, C.; McDonagh, T.; Miller, C.A. Treatment Adherence in a Randomized Controlled Trial of Pirfenidone in HFpEF: Determinants and Impact on Efficacy. J. Card. Fail. 2023, 29, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, Y.; Chen, Z.; Ma, Q.; Abd-Elhamid, A.I.; Feng, B.; Sun, B.; Wu, J. Biomimetic cardiac fibrotic model for anti-fibrotic drug screening. Tissue Eng. Part C Methods 2023, 29, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M.; Shull, J.G.; Vicens-Zygmunt, V.; Rivera-Ortega, P.; Antoniou, K.; Bonella, F.; Renzoni, E.; Russell, A.M.; Maher, T.M.; Vancheri, A.; et al. Gastrointestinal pirfenidone adverse events in idiopathic pulmonary fibrosis depending on diet: The MADIET clinical trial. Eur. Respir. J. 2023, 62, 2300262. [Google Scholar] [CrossRef] [PubMed]
- Alexanian, M.; Haldar, S.M. The Cardiac Myofibroblast. Circ. Res. 2018, 123, 1258–1260. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- De Rosa, S.; Chiefari, E.; Salerno, N.; Ventura, V.; D’Ascoli, G.L.; Arcidiacono, B.; Ambrosio, G.; Bilotta, F.L.; Torella, D.; Foti, D.; et al. HMGA1 is a novel candidate gene for myocardial infarction susceptibility. Int. J. Cardiol. 2017, 227, 331–334. [Google Scholar] [CrossRef]
- Alexanian, M.; Przytycki, P.F.; Micheletti, R.; Padmanabhan, A.; Ye, L.; Travers, J.G.; Gonzalez-Teran, B.; Silva, A.C.; Duan, Q.; Ranade, S.S.; et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 2021, 595, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.N.; Bolli, R.; Hare, J.M. Clinical Studies of Cell Therapy in Cardiovascular Medicine: Recent Developments and Future Directions. Circ. Res. 2018, 123, 266–287. [Google Scholar] [CrossRef]
- Fernandez-Aviles, F.; Sanz-Ruiz, R.; Climent, A.M.; Badimon, L.; Bolli, R.; Charron, D.; Fuster, V.; Janssens, S.; Kastrup, J.; Kim, H.S.; et al. Global position paper on cardiovascular regenerative medicine. Eur. Heart J. 2017, 38, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.K.; Rhee, J.W.; Wu, J.C. Adult Stem Cell Therapy and Heart Failure, 2000 to 2016: A Systematic Review. JAMA Cardiol. 2016, 1, 831–841. [Google Scholar] [CrossRef]
- Nam, Y.J.; Song, K.; Luo, X.; Daniel, E.; Lambeth, K.; West, K.; Hill, J.A.; DiMaio, J.M.; Baker, L.A.; Bassel-Duby, R.; et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. USA 2013, 110, 5588–5593. [Google Scholar] [CrossRef]
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Ieda, M. Cardiac regeneration by direct reprogramming in this decade and beyond. Inflamm. Regen. 2021, 41, 20. [Google Scholar] [CrossRef]
- Christman, K.L.; Vardanian, A.J.; Fang, Q.; Sievers, R.E.; Fok, H.H.; Lee, R.J. Injectable fibrin scaffold improves cell transplant survival, reduces infarct expansion, and induces neovasculature formation in ischemic myocardium. J. Am. Coll. Cardiol. 2004, 44, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Christman, K.L.; Fok, H.H.; Sievers, R.E.; Fang, Q.; Lee, R.J. Fibrin glue alone and skeletal myoblasts in a fibrin scaffold preserve cardiac function after myocardial infarction. Tissue Eng. 2004, 10, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Janbandhu, V.; Tallapragada, V.; Patrick, R.; Li, Y.; Abeygunawardena, D.; Humphreys, D.T.; Martin, E.; Ward, A.O.; Contreras, O.; Farbehi, N.; et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell 2022, 29, 281–297.e12. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.M.; Mesfin, J.M.; Hunter, J.; Cattaneo, P.; Guimaraes-Camboa, N.; Braden, R.L.; Luo, C.; Hill, R.C.; Dzieciatkowska, M.; Hansen, K.C.; et al. Myocardial matrix hydrogel acts as a reactive oxygen species scavenger and supports a proliferative microenvironment for cardiomyocytes. Acta Biomater. 2022, 152, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Odstrcil, M.S.; Lee, C.J.; Sobieski, C.; Weisdorf, D.; Couriel, D. Access to CAR T-cell therapy: Focus on diversity, equity and inclusion. Blood Rev. 2023, 63, 101136. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Rurik, J.G.; Tombacz, I.; Yadegari, A.; Mendez Fernandez, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Kuppe, C.; Ramirez Flores, R.O.; Li, Z.; Hayat, S.; Levinson, R.T.; Liao, X.; Hannani, M.T.; Tanevski, J.; Wunnemann, F.; Nagai, J.S.; et al. Spatial multi-omic map of human myocardial infarction. Nature 2022, 608, 766–777. [Google Scholar] [CrossRef]
- Richards, D.J.; Li, Y.; Kerr, C.M.; Yao, J.; Beeson, G.C.; Coyle, R.C.; Chen, X.; Jia, J.; Damon, B.; Wilson, R.; et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 2020, 4, 446–462. [Google Scholar] [CrossRef]
- Hofbauer, P.; Jahnel, S.M.; Papai, N.; Giesshammer, M.; Deyett, A.; Schmidt, C.; Penc, M.; Tavernini, K.; Grdseloff, N.; Meledeth, C.; et al. Cardioids reveal self-organizing principles of human cardiogenesis. Cell 2021, 184, 3299–3317.e22. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiovascular Disease | Model | Molecular Mechanisms | Main Outcome | References |
|---|---|---|---|---|
| TAC-induced cardiac pressure overload | C57BL/6j and CCR2−/− mice | Increased release of TGF-β1 and IL-10 from cardiac macrophages in TAC mice | Myofibroblast differentiation and collagen production | [45] |
| Cardiac fibrosis with diastolic dysfunction | Patients with hypertension and HFpEF; 8–30 wk-old CCR2−/−, CX3CR1CreER and IL10fl/fl C57BL/6 mice | IL-10 contributes to a macrophage phenotype shift toward a profibrotic subset, which activates fibroblasts | Cardiac fibrosis with diastolic dysfunction | [60] |
| Inflammatory dilated cardiomyopathy (DCMi) | 6–10 wk-old WT, IL17ra−/− BALB/cJ and CBy.PL(B6)-Thy1a/ScrJ (Thy1.1) founder mice | IL-17A induces the production of GM-CSF by CFs, leading to infiltration of Ly6Chigh MO/MΦs | IL-17A directs the conversion of Ly6Chigh MO/MΦ trans to a more pro-inflammatory phenotype via CF-derived GM-CSF | [64] |
| MI | 6–8 wk-old C57BL/6 mice and Trib1–/– mice of a mixed background of C57BL/6 and SV129 | IL-4 treatment increased the number of cardiac M2-like macrophages, which increased the activation of CFs | IL-4 is a potential biological drug for treating MI | [65] |
| MI | 7–10 wk-old female WKY rats | CDCs reduce the number of CD68+ macrophages within the ischemic heart | CDC limits acute injury and attenuates cardiac fibrosis | [66] |
| MI | 6–8 wk-old female WKY rats | Reduced levels of IL-1β and TNF-α in the peri-infarct region | CSps enhance cardiomyocyte proliferation and angiogenesis and attenuate hypertrophy and fibrosis | [67] |
| AMI | Yucatan mini-pigs | / | IC delivery of allo-CDCs is safe, feasible, and effective in cardioprotection, reducing IS, preventing MVO, and attenuating adverse acute remodeling | [68] |
| MI | 8–10 wk-old WT male C57BL/6 and MMP12−/− mice | Significantly increased mRNA expression of CXCL1, CXCL2, and CXCL5 in MMP12−/− mice | MMP-12 production by Ly6Clow macrophages promotes wound healing | [69] |
| Molecules | Cellular Origin | Molecular Mechanisms | References |
|---|---|---|---|
| TGF-β | Macrophages, cardiomyocytes, and fibroblasts themselves | Activation of downstream SMAD3 via TGF-β receptor 1/ALK5 in fibroblasts induced activated fibroblasts to express αSMA, collagen I, Comp, periosteal proliferator protein, and CTGF | [61] |
| IL-4 | / | Inhibition of the increase in the number of M2-like macrophages and increase in the activation level of fibroblasts improve the prognosis of MI | [65,70] |
| IL-6 | Macrophages and fibroblasts | IL-6 acts as a downstream signal for HIMF and activates the MAPK and CaMKII-STAT3 pathways | [71] |
| IL-17A | / | IL-17A induces the production of chemokines by CFs, leading to an infiltration of neutrophils and Ly6Chigh MO/MΦs in the heart | [64] |
| MMP-2, MMP-9, MMP-12 | Neutrophils and macrophages | MMP-2 and MMP-9 over-grade the ECM in the early stages of MI. MMP-12−/− mice show increased neutrophil numbers, upregulated MMP-9, and reduced fibrosis and myofibroblast numbers | [69,72] |
| CX3CR 1 | Macrophages | Altered activity of CFs, resulting in decreased ECM content in the marginal zone and increased cardiac contractility | [73] |
| microRNA-21 | Macrophages | MicroRNA-21 inhibits ERK signaling and enhances cardiac fibroblast survival by suppressing the expression of SPRY1 in CFs | [74] |
| microRNA-155 | Macrophages and fibroblasts | MicroRNA-155 inhibits cardiac fibroblast proliferation by downregulating Sos1 expression and promotes inflammation by decreasing cytokine signaling inhibitor 1 expression | [75] |
| TLR2 | Macrophages | TLR2 deficiency inhibits macrophage-dependent CF activation via modulation of the TGF-β/Smad2/3 pathway | [76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Qiao, Y.; Yan, D.; Meng, Q. Targeting Interactions between Fibroblasts and Macrophages to Treat Cardiac Fibrosis. Cells 2024, 13, 764. https://doi.org/10.3390/cells13090764
Yang B, Qiao Y, Yan D, Meng Q. Targeting Interactions between Fibroblasts and Macrophages to Treat Cardiac Fibrosis. Cells. 2024; 13(9):764. https://doi.org/10.3390/cells13090764
Chicago/Turabian StyleYang, Bo, Yan Qiao, Dong Yan, and Qinghang Meng. 2024. "Targeting Interactions between Fibroblasts and Macrophages to Treat Cardiac Fibrosis" Cells 13, no. 9: 764. https://doi.org/10.3390/cells13090764
APA StyleYang, B., Qiao, Y., Yan, D., & Meng, Q. (2024). Targeting Interactions between Fibroblasts and Macrophages to Treat Cardiac Fibrosis. Cells, 13(9), 764. https://doi.org/10.3390/cells13090764