
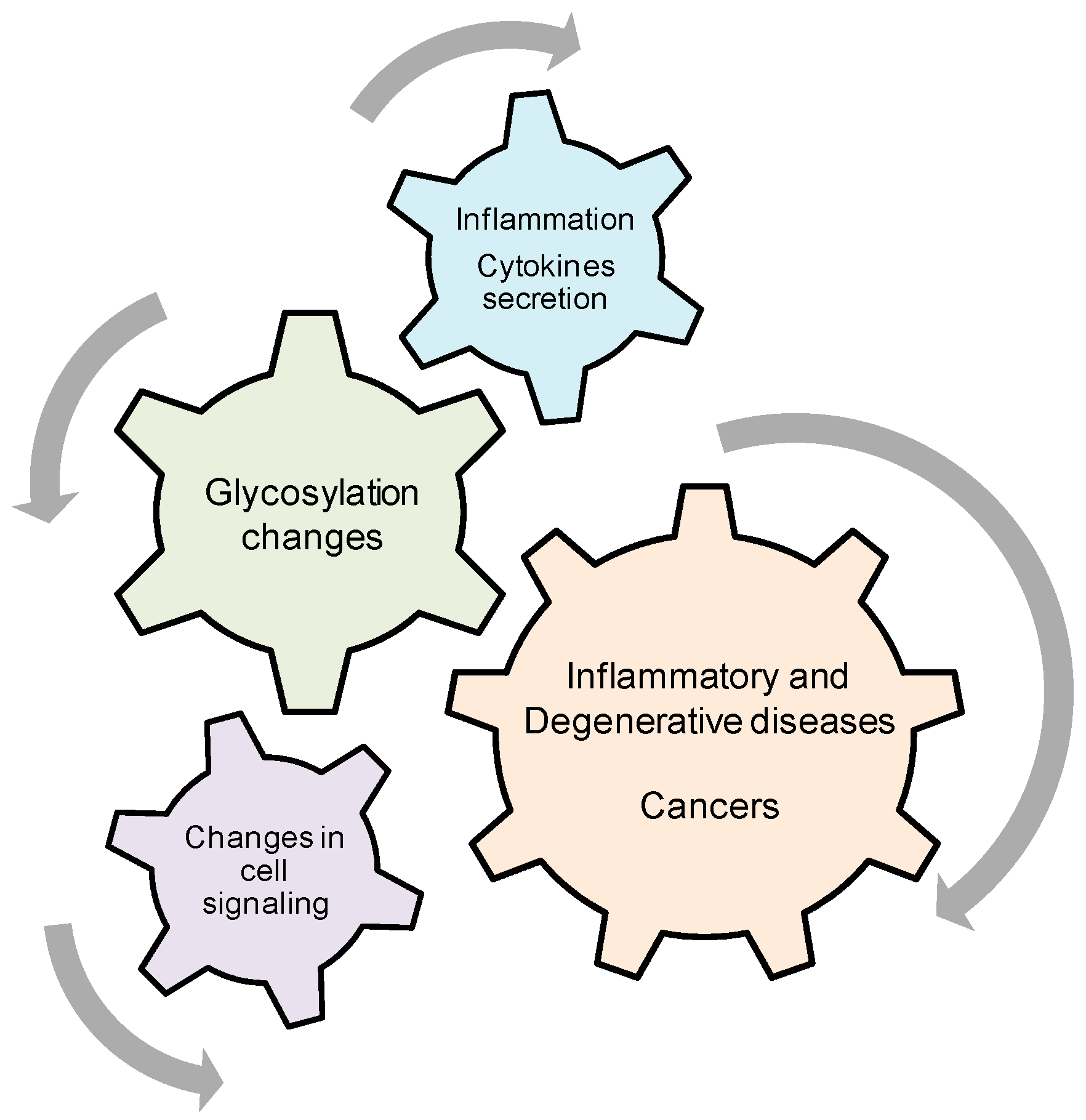
Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regulation of Mucin O-Glycosylation by Pro-Inflammatory Cytokines
2.1. Glycosylation and Sulfation Alterations of Mucin Type O-Glycans in Cystic Fibrosis
2.1.1. Physiopathology of Cystic Fibrosis
2.1.2. Biosynthesis of Human Bronchial Mucins and Their Alterations in CF
2.1.2.1. Bronchial Mucin Biosynthesis
2.1.2.2. Alterations of Mucin Glycosylation and Sulfation of CF: Influence on P. aeruginosa Adhesion
2.2. Inflammation in CF and Altered Mucin Glycosylation
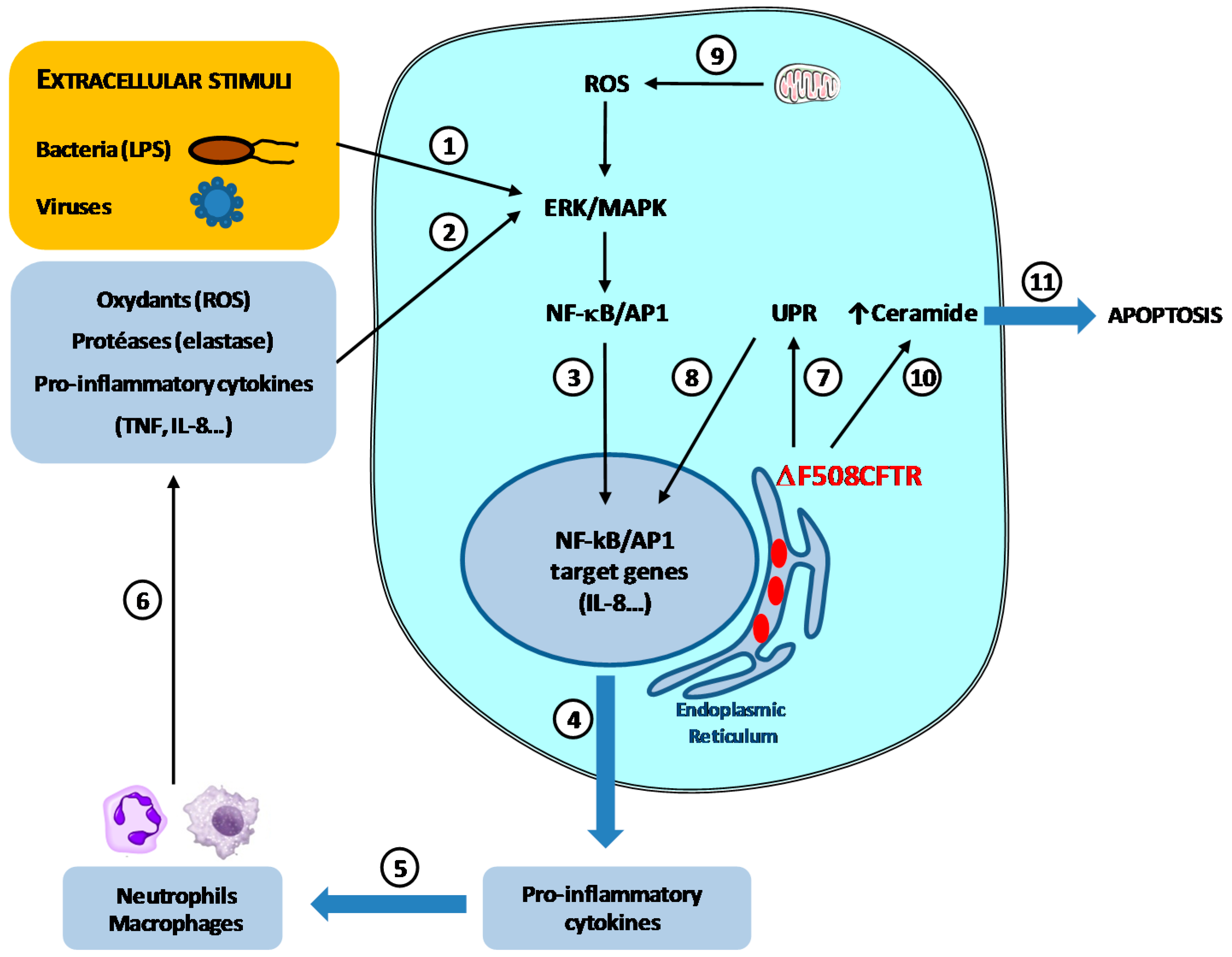
2.2.1. The Vicious Circle of Inflammation/Infection in CF Airways
2.2.2. Influence of Pro-Inflammatory Cytokines on the Expression and Activity of GTs and SulfoTs
2.3. Signaling Pathways Involved in the Regulation of sLex Biosynthesis by TNF in the Human Bronchial Mucosa; Relation with P. aeruginosa Adhesion
2.4. Mucins in Inflammatory Bowel Diseases
3. Regulation of Ganglioside Expression by Pro-Inflammatory Cytokines
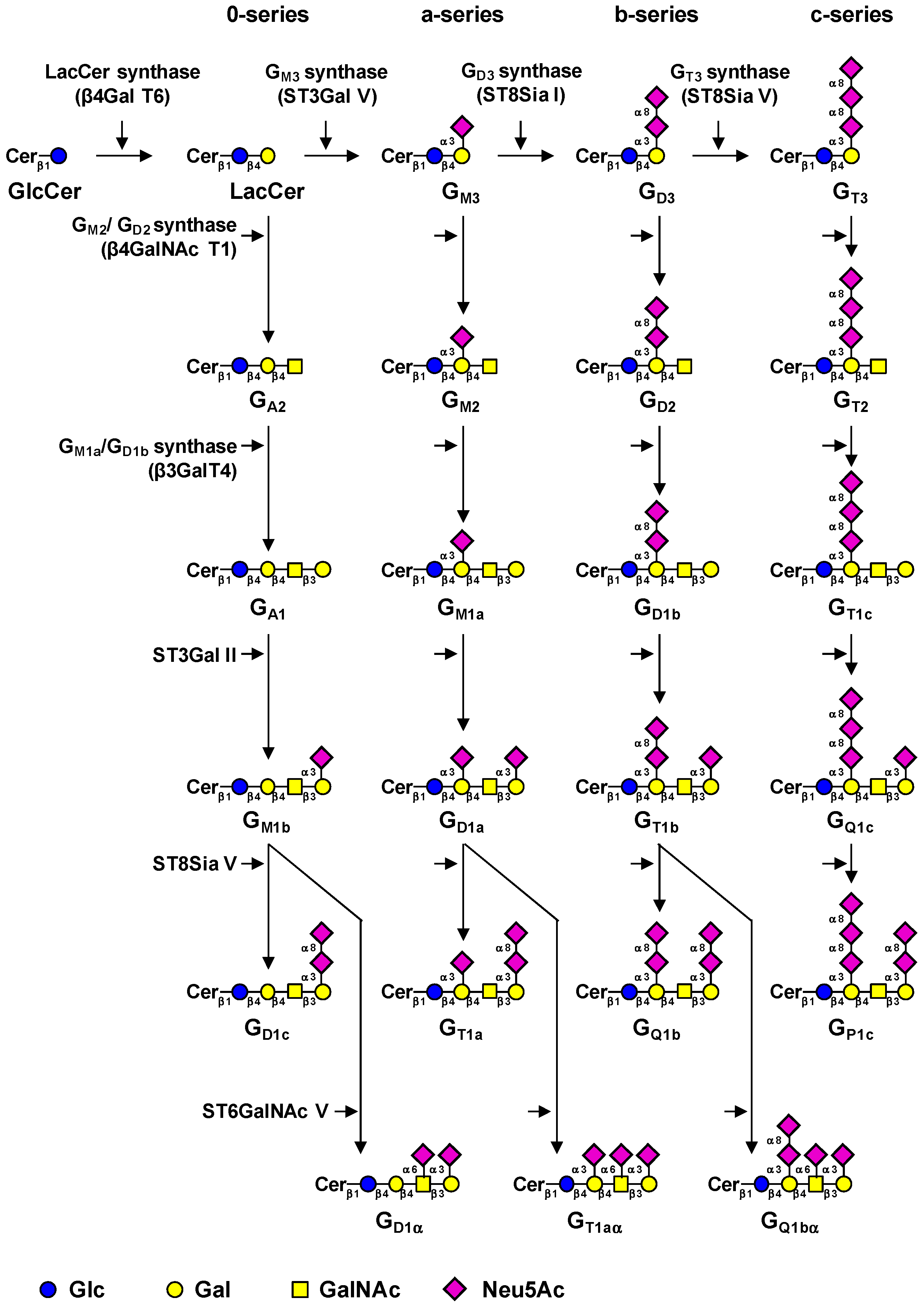
3.1. Biosynthesis of Gangliosides
3.2. Regulation of Ganglioside-Specific Glycosyltransferases by Pro-Inflammatory Cytokines
3.2.1. Regulation of Ganglioside Expression During Inflammatory Reactions may be Involved in the Development of Cancers
3.2.2. Regulatory Function of GSLs in Inflammation and Neurodegeneration
3.3. Effect of Cell Surface Gangliosides on the Activation of Receptors Tyrosine Kinases and Cell Signaling
3.3.1. Monosialogangliosides Are Negative Sensors of RTKs Signaling
3.3.2. Disialogangliosides as Activators of RTKs Signaling
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kim, Y.; Varki, A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj. J. 1997, 14, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.; Läubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.I.; Adachi, Y.; Curiel, D.T.; Kawashima, R.; Kannagi, R.; Nishimoto, N.; Dohi, T. Therapeutic adenoviral gene transfer of a glycosyltransferase for prevention of peritoneal dissemination and metastasis of gastric cancer. Cancer Gene Ther. 2014, 21, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Mizuguchi, S.; Izumi, N.; Chung, K.; Hanada, S.; Inoue, H.; Suehiro, S.; Nishiyama, N. Sialyl Lewis X as a predictor of skip N2 metastasis in clinical stage IA non-small cell lung cancer. World J. Surg. Oncol. 2013, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Gakhar, G.; Navarro, V.N.; Jurish, M.; Lee, G.Y.; Tagawa, S.T.; Akhtar, N.H.; Seandel, M.; Geng, Y.; Liu, H.; Bander, N.H.; et al. Circulating tumor cells from prostate cancer patients interact with E-selectin under physiologic blood flow. PLoS ONE 2013, 8, e85143. [Google Scholar] [CrossRef] [PubMed]
- Harduin-Lepers, A.; Krzewinski-Recchi, M.A.; Colomb, F.; Foulquier, F.; Groux-Degroote, S.; Delannoy, P. Sialyltransferases functions in cancers. Front. Biosci. (Elite Ed.) 2012, 4, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, W.; Mackiewicz, A. Interleukin-6-type cytokine-induced changes in acute phase protein glycosylation. Ann. N. Y. Acad. Sci. 1995, 762, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Degroote, S.; Lafitte, J.J.; Lamblin, G.; Perini, J.M.; Roussel, P. Tumor necrosis factor alpha increases the expression of glycosyltransferases and sulfotransferases responsible for the biosynthesis of sialylated and/or sulfated Lewis x epitopes in the human bronchial mucosa. J. Biol. Chem. 2002, 277, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Krzewinski-Recchi, M.A.; Cazet, A.; Vincent, A.; Lehoux, S.; Lafitte, J.J.; van Seuningen, I.; Delannoy, P. IL-6 and IL-8 increase the expression of glycosyltransferases and sulfotransferases involved in the biosynthesis of sialylated and/or sulfated Lewis x epitopes in the human bronchial mucosa. Biochem. J. 2008, 410, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Bobowski, M.; Steenackers, A.; Le Bourhis, X.; Delannoy, P. How Do Gangliosides Regulate RTKs Signaling? Cells 2013, 2, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Guérardel, Y.; Groux-Degroote, S.; Delannoy, P. Gangliosphyngolipids: Their structure and biological roles. In Carbohydrate Chemistry: State-of-the-Art and Challenges for Drug Development; Cipolla, L., Ed.; Imperial College Press: London, UK, 2015; pp. 35–56. [Google Scholar]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Galietta, L.J. Targeting ion channels in cystic fibrosis. J. Cyst. Fibros. 2015, 14, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Mutation Database. Available online: http://www.genet.sickkids.on.ca/cftr (accessed on 21 September 2016).
- Lakshmanan, I.; Ponnusamy, M.P.; Macha, M.A.; Haridas, D.; Majhi, P.D.; Kaur, S.; Jain, M.; Batra, S.K.; Ganti, A.K. Mucins in lung cancer: Diagnostic, prognostic, and therapeutic implications. J. Thorac. Oncol. 2015, 10, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Hauber, H.P.; Foley, S.C.; Hamid, Q. Mucin overproduction in chronic inflammatory lung disease. Can. Respir. J. 2006, 13, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.L.; Sloane, A.J.; Robinson, L.J.; Prasad, S.S.; Lindner, R.A.; Robinson, M.; Bye, P.T.; Nielson, D.W.; Harry, J.L.; Packer, N.H.; et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology 2007, 17, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Copin, M.C.; Buisine, M.P.; Devisme, L.; Leroy, X.; Escande, F.; Gosselin, B.; Aubert, J.P.; Porchet, N. Normal respiratory mucosa, precursor lesions and lung carcinomas: Differential expression of human mucin genes. Front. Biosci. 2001, 6, D1264–D1275. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.M.; Koo, J.S. Airway mucus: The good, the bad, the sticky. Pharmacol. Ther. 2009, 121, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, K.T.; Clausen, H. Site-specific protein O-glycosylation modulates proprotein processing—Deciphering specific functions of the large polypeptide GalNAc-transferase gene family. Biochim. Biophys. Acta 2012, 1820, 2079–2094. [Google Scholar] [CrossRef] [PubMed]
- Lo-Guidice, J.M.; Wieruszeski, J.M.; Lemoine, J.; Verbert, A.; Roussel, P.; Lamblin, G. Sialylation and sulfation of the carbohydrate chains in respiratory mucins from a patient with cystic fibrosis. J. Biol. Chem. 1994, 269, 18794–18813. [Google Scholar] [PubMed]
- Brockhausen, I.; Schachter, H.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 9. [Google Scholar]
- Boat, T.F.; Kleinerman, J.I.; Fanaroff, A.A.; Stern, R.C. Human tracheobronchial secretions: Development of mucous glycoprotein and lysozyme-secreting systems. Pediatr. Res. 1977, 11, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Wesley, A.; Forstner, J.; Qureshi, R.; Mantle, M.; Forstner, G. Human intestinal mucin in cystic fibrosis. Pediatr. Res. 1983, 17, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Carnoy, C.; Ramphal, R.; Scharfman, A.; Lo-Guidice, J.M.; Houdret, N.; Klein, A.; Galabert, C.; Lamblin, G.; Roussel, P. Altered carbohydrate composition of salivary mucins from patients with cystic fibrosis and the adhesion of Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 1993, 9, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Lamblin, G.; Boersma, A.; Lhermitte, M.; Roussel, P.; Mutsaers, J.H.; van Halbeek, H.; Vliegenthart, J.F. Further characterization, by a combined high-performance liquid chromatography/1H-NMR approach, of the heterogeneity displayed by the neutral carbohydrate chains of human bronchial mucins. Eur. J. Biochem. 1984, 143, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Royall, J.A.; Damera, G.; Sachdev, G.P.; Cummings, R.D. Altered O-glycosylation and sulfation of airway mucins associated with cystic fibrosis. Glycobiology 2005, 15, 747–775. [Google Scholar] [CrossRef] [PubMed]
- Davril, M.; Degroote, S.; Humbert, P.; Galabert, C.; Dumur, V.; Lafitte, J.J.; Lamblin, G.; Roussel, P. The sialylation of bronchial mucins secreted by patients suffering from cystic fibrosis or from chronic bronchitis is related to the severity of airway infection. Glycobiology 1999, 9, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, A.; Degroote, S.; Beau, J.; Lamblin, G.; Roussel, P.; Mazurier, J. Pseudomonas aeruginosa binds to neoglycoconjugates bearing mucin carbohydrate determinants and predominantly to sialyl-Lewis x conjugates. Glycobiology 1999, 9, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, A.; Arora, S.K.; Delmotte, P.; van Brussel, E.; Mazurier, J.; Ramphal, R.; Roussel, P. Recognition of Lewis x derivatives present on mucins by flagellar components of Pseudomonas aeruginosa. Infect. Immun. 2001, 69, 5243–5248. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Stoykova, L.I.; Trindade, A.J.; Glick, M.C.; Scanlin, T.F. Altered terminal glycosylation and the pathophysiology of CF lung disease. J. Cyst. Fibros. 2004, 3, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Panuska, J.R.; Konstan, M.W.; Hilliard, K.A.; Hilliard, J.B.; Ghnaim, H.; Berger, M. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 1995, 152, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.; Henry, A.; Kreindler, J.L.; Dubin, P.J.; Ulrich, L.; Steele, C.; Finder, J.D.; Pilewski, J.M.; Carreno, B.M.; Goldman, S.J.; et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: Implications for airway inflammation in cystic fibrosis. J. Immunol. 2005, 175, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Konstan, M.W.; Berger, M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J. Allergy Clin. Immunol. 1999, 104, 72–78. [Google Scholar] [CrossRef]
- Conese, M.; Assael, B.M. Bacterial infections and inflammation in the lungs of cystic fibrosis patients. Pediatr. Infect. Dis. J. 2001, 20, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Corvol, H.; Fitting, C.; Chadelat, K.; Jacquot, J.; Tabary, O.; Boule, M.; Cavaillon, J.M.; Clement, A. Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L997–L1003. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.J.; Soong, G.; Bryan, R.; Saba, S.; Prince, A. Activation of NF-kappaB in airway epithelial cells is dependent on CFTR trafficking and Cl− channel function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L71–L78. [Google Scholar] [PubMed]
- Velsor, L.W.; van Heeckeren, A.; Day, B.J. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L31–L38. [Google Scholar] [PubMed]
- Velsor, L.W.; Kariya, C.; Kachadourian, R.; Day, B.J. Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Respir. Cell Mol. Biol. 2006, 35, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Brodlie, M.; McKean, M.C.; Johnson, G.E.; Gray, J.; Fisher, A.J.; Corris, P.A.; Lordan, J.L.; Ward, C. Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2010, 182, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Teichgräber, V.; Ulrich, M.; Endlich, N.; Riethmüller, J.; Wilker, B.; de Oliveira-Munding, C.C.; van Heeckeren, A.M.; Barr, M.L.; von Kürthy, G.; Schmid, K.W.; et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008, 14, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, C.; Remouchamps, C.; Hennuy, B.; Vanderplasschen, A.; Chariot, A.; Tabruyn, S.P.; Oury, C.; Bours, V. Role of IKK and ERK pathways in intrinsic inflammation of cystic fibrosis airways. Biochem. Pharmacol. 2007, 73, 1982–1994. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cymberknoh, M.; Kerem, E.; Ferkol, T.; Elizur, A. Airway inflammation in cystic fibrosis: Molecular mechanisms and clinical implications. Thorax 2013, 68, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Degroote, S.; Merten, M.; Bernigaud, A.; van Seuningen, I.; Figarella, C.; Roussel, P.; Perini, J.M. Influence of culture conditions on the α1,2-fucosyltransferase and MUC gene expression of a transformed cell line MM-39 derived from human tracheal gland cells. Biochimie 2001, 83, 749–755. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell. Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Inouye, Y.; Okano, T.; Taniguchi, A. Regulation of sialyl-Lewis x epitope expression by TNF-alpha and EGF in an airway carcinoma cell line. Glycoconj. J. 2005, 22, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Colomb, F.; Vidal, O.; Bobowski, M.; Krzewinski-Recchi, M.A.; Harduin-Lepers, A.; Mensier, E.; Jaillard, S.; Lafitte, J.J.; Delannoy, P.; Groux-Degroote, S. TNF induces the expression of the sialyltransferase ST3Gal IV in human bronchial mucosa via MSK1/2 protein kinases and increases FliD/sialyl-Lewis(x)-mediated adhesion of Pseudomonas aeruginosa. Biochem. J. 2014, 457, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Colomb, F.; Krzewinski-Recchi, M.A.; El Machhour, F.; Mensier, E.; Jaillard, S.; Steenackers, A.; Harduin-Lepers, A.; Lafitte, J.J.; Delannoy, P.; Groux-Degroote, S. TNF regulates sialyl-Lewisx and 6-sulfo-sialyl-Lewisx expression in human lung through up-regulation of ST3GAL4 transcript isoform BX. Biochimie 2012, 94, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, J.L.; Jia, J.; Choi, W.; Choe, S.; Miao, J.; Xu, Y.; Powell, R.; Lin, J.; Kuang, Z.; Gaskins, H.R.; et al. Pseudomonas aeruginosa pyocyanin modulates mucin glycosylation with sialyl-Lewis(x) to increase binding to airway epithelial cells. Mucosal Immunol. 2016, 9, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Enss, M.L.; Cornberg, M.; Wagner, S.; Gebert, A.; Henrichs, M.; Eisenblätter, R.; Beil, W.; Kownatzki, R.; Hedrich, H.J. Proinflammatory cytokines trigger MUC gene expression and mucin release in the intestinal cancer cell line LS180. Inflamm. Res. 2000, 49, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Sartor, R.B.; Tennyson, G.S.; Riddell, R.H. Experimental models of inflammatory bowel disease. Gastroenterology 1995, 109, 1344–1367. [Google Scholar] [CrossRef]
- Dkhil, M.A.; Delic, D.; Al-Quraishy, S. Goblet cells and mucin related gene expression in mice infected with Eimeria papillata. Sci. World J. 2013, 2013, 439865. [Google Scholar] [CrossRef] [PubMed]
- Pullan, R.D.; Thomas, G.A.; Rhodes, M.; Newcombe, R.G.; Williams, G.T.; Allen, A.; Rhodes, J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 1994, 35, 353–359. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.A.; Horton, L.W.; Mee, A.S. Mucin depletion in inflammatory bowel disease. J. Clin. Pathol. 1990, 43, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Raouf, A.H.; Tsai, H.H.; Parker, N.; Hoffman, J.; Walker, R.J.; Rhodes, J.M. Sulfation of colonic mucin in ulcerative colitis and Crohn’s disease. Clin. Sci. 1992, 83, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.; Tsai, H.H.; Ryder, S.D.; Raouf, A.H.; Rhodes, J.M. Increased rate of sialylation of colonic mucin by cultured ulcerative colitis mucosal explants. Digestion 1995, 56, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.M.; Karlsson, H.; Crespo, J.G.; Johansson, M.E.; Eklund, L.; Sjövall, H.; Hansson, G.C. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 2011, 17, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J. The Role of Sialyl-Tn in Cancer. Int. J. Mol. Sci. 2016, 17, 275. [Google Scholar] [CrossRef] [PubMed]
- Bodger, K.; Halfvarson, J.; Dodson, A.R.; Campbell, F.; Wilson, S.; Lee, R.; Lindberg, E.; Järnerot, G.; Tysk, C.; Rhodes, J.M. Altered colonic glycoprotein expression in unaffected monozygotic twins of inflammatory bowel disease patients. Gut 2006, 55, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Velcich, A.; Yang, W.; Heyer, J.; Fragale, A.; Nicholas, C.; Viani, S.; Kucherlapati, R.; Lipkin, M.; Yang, K.; Augenlicht, L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2002, 295, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wei, B.; Wen, T.; Johansson, M.E.; Liu, X.; Bradford, E.; Thomsson, K.A.; McGee, S.; Mansour, L.; Tong, M.; et al. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J. Clin. Investig. 2011, 121, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.; Liu, X.; Zhao, Y.; Gao, N.; Wu, Q.; Song, K.; Cui, Y.; Li, Y.; McDaniel, J.M.; McGee, S.; et al. Defective Intestinal Mucin-Type O-Glycosylation Causes Spontaneous Colitis-Associated Cancer in Mice. Gastroenterology 2016, 151, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.; Fu, J.; Johansson, M.E.; Liu, X.; Gao, N.; Wu, Q.; Song, J.; McDaniel, J.M.; McGee, S.; Chen, W.; et al. Core 1- and 3-derived O-glycans collectively maintain the colonic mucus barrier and protect against spontaneous colitis in mice. Mucosal Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S.I. The glycosynapse. Proc. Natl. Acad. Sci. USA 2002, 99, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Regina-Todeschini, A.; Todeschini, R.A.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Birkle, S.; Zeng, G.; Gao, L.; Yu, R.K.; Aubry, J. Role of tumor-associated gangliosides in cancer progression. Biochimie 2003, 85, 455–463. [Google Scholar] [CrossRef]
- Prokazova, N.V.; Bergelson, L.D. Gangliosides and atherosclerosis. Lipids 1994, 29, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ariga, T.; McDonald, M.P.; Yu, R.K. Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease—A review. J. Lipid Res. 2008, 49, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Hamamura, K.; Aixinjueluo, W.; Furukawa, K. Biosignals modulated by tumor-associated carbohydrate antigens: Novel targets for cancer therapy. Ann. N. Y. Acad. Sci. 2006, 1086, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Krengel, U.; Bousquet, P.A. Molecular recognition of gangliosides and their potential for cancer immunotherapies. Front. Immunol. 2014, 5, 325. [Google Scholar] [CrossRef] [PubMed]
- Rabu, C.; McIntosh, R.; Jurasova, Z.; Durrant, L. Glycans as targets for therapeutic antitumor antibodies. Future Oncol. 2012, 8, 943–960. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 4638–4643. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Inokuchi, J.; Jimbo, M.; Shimeno, H.; Nagamatsu, A.; Shayman, J.A.; Shukla, G.S.; Radin, N.S. Improved inhibitors of glucosylceramide synthase. J. Biochem. 1992, 111, 191–196. [Google Scholar] [PubMed]
- Nomura, T.; Takizawa, M.; Aoki, J.; Arai, H.; Inoue, K.; Wakisaka, E.; Yoshizuka, N.; Imokawa, G.; Dohmae, N.; Takio, K.; et al. Purification, cDNA cloning, and expression of UDP-Gal: Glucosylceramide β-1,4-galactosyltransferase from rat brain. J. Biol. Chem. 1998, 273, 13570–13577. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, M.; Nomura, T.; Wakisaka, E.; Yoshizuka, N.; Aoki, J.; Arai, H.; Inoue, K.; Hattori, M.; Matsuo, N. cDNA cloning and expression of human lactosylceramide synthase. Biochim. Biophys. Acta 1999, 1438, 301–304. [Google Scholar] [CrossRef]
- Zeng, G.; Yu, R.K. Cloning and transcriptional regulation of genes responsible for synthesis of gangliosides. Curr. Drug Targets 2008, 9, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Ohta, M.; Watanabe, Y.; Matsuda, K.; Ishiyama, K.; Sakoe, K.; Nakamura, M.; Inokuchi, J.; Sanai, Y.; Saito, M. Expression cloning and functional characterization of human cDNA for ganglioside GM3 synthase. J. Biol. Chem. 1998, 273, 31652–31655. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, M.; Yamashiro, S.; Yamamoto, A.; Furukawa, K.; Takamiya, K.; Lloyd, K.O.; Shiku, H.; Furukawa, K. Isolation of GD3 synthase gene by expression cloning of GM3 α-2,8-sialyltransferase cDNA using anti-GD2 monoclonal antibody. Proc. Natl. Acad. Sci. USA 1994, 91, 10455–10459. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Fukuda, M.N.; Hirabayashi, Y.; Kanamori, A.; Sasaki, K.; Nishi, T.; Fukuda, M. Expression cloning of a human GT3 synthase. GD3 and GT3 are synthesized by a single enzyme. J. Biol. Chem. 1996, 271, 3684–3691. [Google Scholar] [PubMed]
- Kim, Y.J.; Kim, K.S.; Do, S.; Kim, C.H.; Kim, S.K.; Lee, Y.C. Molecular cloning and expression of human α-2,8-sialyltransferase (hST8Sia V). Biochem. Biophys. Res. Commun. 1997, 235, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L. Ganglioside designation. Adv. Exp. Med. Biol. 1980, 125, 11. [Google Scholar] [PubMed]
- Bobowski, M.; Cazet, A.; Steenackers, A.; Delannoy, P. Role of complex gangliosides in cancer progression. Carbohydr. Chem. 2012, 37, 1–20. [Google Scholar] [CrossRef]
- Nagata, Y.; Yamashiro, S.; Yodoi, J.; Lloyd, K.O.; Shiku, H.; Furukawa, K. Expression cloning of beta-1,4-N-acetylgalactosaminyltransferase cDNAs that determine the expression of GM2 and GD2 gangliosides. J. Biol. Chem. 1992, 267, 12082–12089. [Google Scholar] [PubMed]
- Amado, M.; Almeida, R.; Carneiro, F.; Levery, S.B.; Holmes, E.H.; Nomoto, M.; Hollingsworth, M.A.; Hassan, H.; Schwientek, T.; Nielsen, P.A.; et al. A family of human beta3-galactosyltransferases. Characterization of four members of a UDP-galactose: β-N-acetylglucosamine/β-N-acetyl-galactosamine/β-1,3-galactosyltransferase family. J. Biol. Chem. 1998, 273, 12770–12778. [Google Scholar] [CrossRef] [PubMed]
- Iber, H.; Zacharias, C.; Sandhoff, K. The c-series gangliosides GT3, GT2, and GP1c are formed in rat liver Golgi by the same set of glycosyltransferases that catalyze the biosynthesis of asialo-, a- and b-series gangliosides. Glycobiology 1992, 2, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, S.; Haraguchi, M.; Furukawa, K.; Takamiya, K.; Yamamoto, A.; Nagata, Y.; Lloyd, K.O.; Shiku, H.; Furukawa, K. Substrate specificity of β-1,4-N-acetylgalactosaminyltransferase in vitro and in cDNA-transfected cells. GM2/GD2 synthase efficiently generates asialo-GM2 in certain cells. J. Biol. Chem. 1995, 270, 6149–6155. [Google Scholar] [PubMed]
- Sturgill, E.R.; Aoki, K.; Lopez, P.H.; Colacurcio, D.; Vajn, K.; Lorenzini, I.; Majic, S.; Yang, W.H.; Heffer, M.; Tiemeyer, M.; et al. Biosynthesis of the major brain gangliosides GD1a and GT1b. Glycobiology 2012, 22, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Okajima, T.; Fukumoto, S.; Ito, H.; Kiso, M.; Hirabayashi, Y.; Urano, T.; Furukawa, K. Molecular cloning of brain-specific GD1α synthase (ST6GalNAc V) containing CAG/Glutamine repeats. J. Biol. Chem. 1999, 274, 30557–30562. [Google Scholar] [CrossRef] [PubMed]
- Hidari, J.K.; Ichikawa, S.; Furukawa, K.; Yamasaki, M.; Hirabayashi, Y. β-1–4-N-acetylgalactosaminyltransferase can synthesize both asialoglycosphingolipid GM2 and glycosphingolipid GM2 in vitro and in vivo: Isolation and characterization of a β-1,4-N-acetylgalactosaminyltransferase cDNA clone from rat ascites hepatoma cell line AH7974F. Biochem. J. 1994, 303, 957–965. [Google Scholar] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Guérardel, Y.; Julien, S.; Delannoy, P. Gangliosides in breast cancer: New perspectives. Biochemistry (Moscow) 2015, 80, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Markotić, A.; Lümen, R.; Marusić, A.; Jonjić, S.; Müthing, J. Ganglioside expression in tissues of mice lacking the tumor necrosis factor receptor 1. Carbohydr. Res. 1999, 321, 75–87. [Google Scholar] [CrossRef]
- Furukawa, K.; Arita, Y.; Satomi, N.; Eisinger, M.; Lloyd, K.O. Tumor necrosis factor enhances GD3 ganglioside expression in cultured human melanocytes. Arch. Biochem. Biophys. 1990, 281, 70–75. [Google Scholar] [CrossRef]
- Van de Kar, N.C.; Monnens, L.A.; van Hinsbergh, V.W. Tumor necrosis factor and interleukin 1 induce expression of the glycolipid verotoxin receptor in human endothelial cells. Implications for the pathogenesis of the haemolytic uraemic syndrome. Behring Inst. Mitt. 1993, 92, 202–209. [Google Scholar]
- Raval, G.; Biswas, S.; Rayman, P.; Biswas, K.; Sa, G.; Ghosh, S.; Thornton, M.; Hilston, C.; Das, T.; Bukowski, R.; et al. TNFα induction of GM2 expression on renal cell carcinomas promotes T cell dysfunction. J. Immunol. 2007, 178, 6642–6652. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, S.; Okada, M.; Haraguchi, M.; Furukawa, K.; Lloyd, K.O.; Shiku, H.; Furukawa, K. Expression of alpha 2,8-sialyltransferase (GD3 synthase) gene in human cancer cell lines: High level expression in melanomas and up-regulation in activated T lymphocytes. Glycoconj. J. 1995, 12, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Ichihara, M.; Tajima, O.; Sobue, S.; Kambe, M.; Sugiura, K.; Furukawa, K.; Furukawa, K. UVB-irradiated keratinocytes induce melanoma-associated ganglioside GD3 synthase gene in melanocytes via secretion of tumor necrosis factor α and interleukin 6. Biochem. Biophys. Res. Commun. 2014, 445, 504–510. [Google Scholar] [CrossRef] [PubMed]
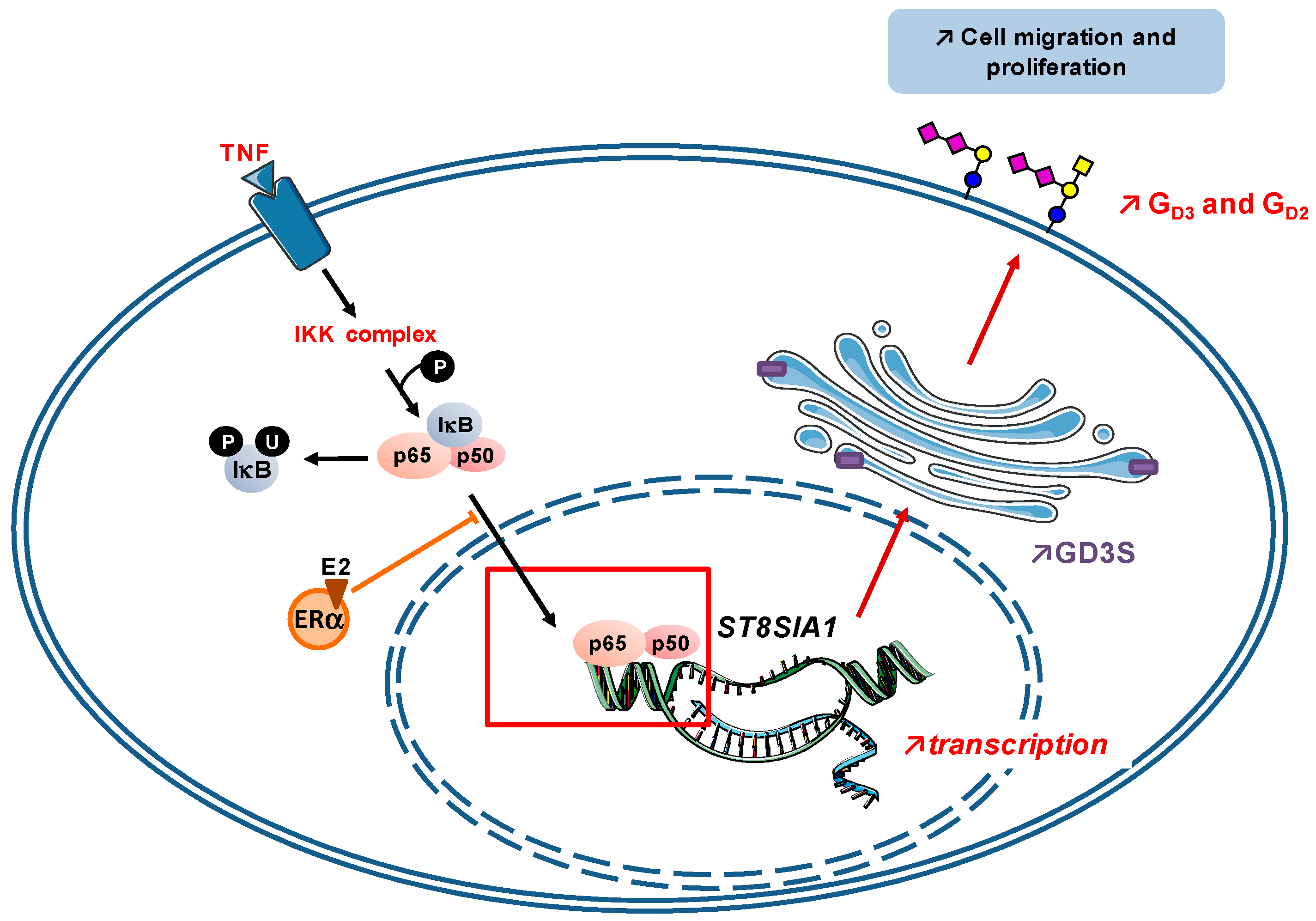
- Kang, N.Y.; Kim, C.H.; Kim, K.S.; Ko, J.H.; Lee, J.H.; Jeong, Y.K.; Lee, Y.C. Expression of the human CMP-NeuAc:GM3 alpha2,8-sialyltransferase (GD3 synthase) gene through the NF-kappaB activation in human melanoma SK-MEL-2 cells. Biochim. Biophys. Acta 2007, 1769, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.Y.; Dae, H.M.; Song, N.R.; Kim, K.S.; Kim, C.H.; Lee, Y.C. Valproic acid induces transcriptional activation of human GD3 synthase (hST8Sia I) in SK-N-BE(2)-C human neuroblastoma cells. Mol. Cells 2009, 27, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Bobowski, M.; Vincent, A.; Steenackers, A.; Colomb, F.; van Seuningen, I.; Julien, S.; Delannoy, P. Estradiol represses the GD3 synthase gene ST8SIA1 expression in human breast cancer cells by preventing NFκB binding to ST8SIA1 promoter. PLoS ONE 2013, 8, e62559. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Tajima, O.; Egashira, N.; Ohmi, Y.; Fukue, Y.; Mishima, K.; Iwasaki, K.; Fujiwara, M.; Sugiura, Y.; Furukawa, K.; Furukawa, K. Dysfunction of muscarinic acetylcholine receptors as a substantial basis for progressive neurological deterioration in GM3-only mice. Behav. Brain Res. 2010, 206, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Ohkawa, Y.; Tajima, O.; Sugiura, Y.; Furukawa, K.; Furukawa, K. Ganglioside deficiency causes inflammation and neurodegeneration via the activation of complement system in the spinal cord. J. Neuroinflamm. 2014, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A.; et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Boukhris, A.; Schule, R.; Loureiro, J.L.; Lourenço, C.M.; Mundwiller, E.; Gonzalez, M.A.; Charles, P.; Gauthier, J.; Rekik, I.; Acosta Lebrigio, R.F.; et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 93, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Fujita, M.; Maeda, Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: Recent progress. J. Biochem. 2008, 144, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Nicola, D.; Doncel-Pérez, E.; Nieto-Sampedro, M. Regulation by GD3 of the proinflammatory response of microglia mediated by interleukin-15. J. Neurosci. Res. 2006, 83, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, Y.; Cao, F.; Qin, Y.; Li, W.; Zhang, J. Ganglioside GD1a suppresses LPS-induced pro-inflammatory cytokines in RAW264.7 macrophages by reducing MAPKs and NF-κB signaling pathways through TLR4. Int. Immunopharmacol. 2015, 28, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Bremer, E.G.; Schlessinger, J.; Hakomori, S.I. Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J. Biol. Chem. 1986, 261, 2434–2440. [Google Scholar] [PubMed]
- Bremer, E.G.; Hakomori, S.I. Gangliosides as receptor modulators. Adv. Exp. Med. Biol. 1984, 174, 381–394. [Google Scholar] [PubMed]
- Hakomori, S.; Igarashi, Y. Functional role of glycosphingolipids in cell recognition and signaling. J. Biochem. 1995, 118, 1091–1103. [Google Scholar] [PubMed]
- Lai, A.Z.; Abella, J.V.; Park, M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol. 2009, 19, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Yoon, S.J.; Freire-de-Lima, L.; Kim, J.H.; Hakomori, S.I. Control of cell motility by interaction of gangliosides, tetraspanins, and epidermal growth factor receptor in A431 versus KB epidermoid tumor cells. Carbohydr. Res. 2009, 344, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, A.R.; Dos Santos, J.N.; Handa, K.; Hakomori, S.I. Ganglioside GM2-tetraspanin CD82 complex inhibits met and its cross-talk with integrins, providing a basis for control of cell motility through glycosynapse. J. Biol. Chem. 2007, 282, 8123–8133. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Ohkawa, Y.; Yamauchi, Y.; Hamamura, K.; Ohmi, Y.; Furukawa, K. Fine tuning of cell signals by glycosylation. J. Biochem. 2012, 151, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Zhang, J.; Xu, Y.; Tian, Y.; Ma, K. Ganglioside GM3 inhibits hepatoma cell motility via down-regulating activity of EGFR and PI3K/AKT signaling pathway. J. Cell. Biochem. 2013, 114, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, X.; Zhang, J.; Li, Y.; Ma, K. Synergistic inhibition of cell migration by tetraspanin CD82 and gangliosides occurs via the EGFR or cMet-activated Pl3K/Akt signalling pathway. Int. J. Biochem. Cell. Biol. 2013, 45, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Coskun, Ü.; Grzybek, M.; Drechsel, D.; Simons, K. Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. USA 2011, 108, 9044–9048. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, B.L.; Clark, S.H.; Zhang, C. Inhibition of human neuroblastoma cell proliferation and EGF receptor phosphorylation by gangliosides GM1, GM3, GD1A and GT1B. Cell Prolif. 2002, 35, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Miljan, E.A.; Meuillet, E.J.; Mania-Farnell, B.; George, D.; Yamamoto, H.; Simon, H.G.; Bremer, E.G. Interaction of the extracellular domain of the epidermal growth factor receptor with gangliosides. J. Biol. Chem. 2002, 277, 10108–10113. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Nakayama, K.; Hikita, T.; Handa, K.; Hakomori, S.I. Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 18987–18991. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, N.; Yoon, S.J.; Itoh, K.; Nakayama, K. Tyrosine kinase activity of epidermal growth factor receptor is regulated by GM3 binding through carbohydrate to carbohydrate interactions. J. Biol. Chem. 2009, 284, 6147–6155. [Google Scholar] [CrossRef] [PubMed]
- Meuillet, E.; Cremel, G.; Dreyfus, H.; Hicks, D. Differential modulation of basic fibroblast and epidermal growth factor receptor activation by ganglioside GM3 in cultured retinal Müller glia. Glia 1996, 17, 206–216. [Google Scholar] [CrossRef]
- Toledo, M.S.; Suzuki, E.; Handa, K.; Hakomori, S. Effect of ganglioside and tetraspanins in microdomains on interaction of integrins with fibroblast growth factor receptor. J. Biol. Chem. 2005, 280, 16227–16234. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, A.R.; Dos Santos, J.N.; Handa, K.; Hakomori, S.I. Ganglioside GM2/GM3 complex affixed on silica nanospheres strongly inhibits cell motility through CD82/cMet-mediated pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Mukherjee, P. Ganglioside GM3 Is Antiangiogenic in Malignant Brain Cancer. J. Oncol. 2010, 2010, 961243. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.W.; Kim, S.J.; Choi, H.J.; Kim, K.J.; Kim, M.J.; Kim, S.H.; Lee, H.J.; Ko, J.H.; Lee, Y.C.; Suzuki, A.; et al. Ganglioside GM3 inhibits VEGF/VEGFR-2-mediated angiogenesis: Direct interaction of GM3 with VEGFR-2. Glycobiology 2009, 19, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Fukumoto, S.; Furukawa, K.; Ichimura, A.; Miyazaki, H.; Kusunoki, S.; Urano, T.; Furukawa, K. Overexpressed GM1 suppresses nerve growth factor (NGF) signals by modulating the intracellular localization of NGF receptors and membrane fluidity in PC12 cells. J. Biol. Chem. 2004, 279, 33368–33378. [Google Scholar] [CrossRef] [PubMed]
- Mitsuda, T.; Furukawa, K.; Fukumoto, S.; Miyazaki, H.; Urano, T.; Furukawa, K. Overexpression of ganglioside GM1 results in the dispersion of platelet-derived growth factor receptor from glycolipid-enriched microdomains and in the suppression of cell growth signals. J. Biol. Chem. 2002, 277, 11239–11246. [Google Scholar] [CrossRef] [PubMed]
- Veracini, L.; Simon, V.; Richard, V.; Schraven, B.; Horejsi, V.; Roche, S.; Benistant, C. The Csk-binding protein PAG regulates PDGF-induced Src mitogenic signaling via GM1. J. Cell Biol. 2008, 182, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, S.; Mutoh, T.; Hasegawa, T.; Miyazaki, H.; Okada, M.; Goto, G.; Furukawa, K.; Urano, T. GD3 synthase gene expression in PC12 cells results in the continuous activation of TrkA and ERK1/2 and enhanced proliferation. J. Biol. Chem. 2000, 275, 5832–5838. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.; Groux-Degroote, S.; Teylaert, B.; Kwon, K.M.; Lehoux, S.; Slomianny, C.; Kim, C.H.; Le Bourhis, X.; Delannoy, P. GD3 synthase overexpression enhances proliferation and migration of MDA-MB-231 breast cancer cells. Biol. Chem. 2009, 390, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.; Bobowski, M.; Rombouts, Y.; Lefebvre, J.; Steenackers, A.; Popa, I.; Guérardel, Y.; Le Bourhis, X.; Tulasne, D.; Delannoy, P. The ganglioside GD2 induces the constitutive activation of c-Met in MDA-MB-231 breast cancer cells expressing the GD3 synthase. Glycobiology 2012, 22, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.; Lefebvre, J.; Adriaenssens, E.; Julien, S.; Bobowski, M.; Grigoriadis, A.; Tutt, A.; Tulasne, D.; Le Bourhis, X.; Delannoy, P. GD3 synthase expression enhances proliferation and tumor growth of MDA-MB-231 breast cancer cells through c-Met activation. Mol. Cancer Res. 2010, 8, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Hyuga, S.; Kawasaki, N.; Hyuga, M.; Ohta, M.; Shibayama, R.; Kawanishi, T.; Yamagata, S.; Yamagata, T.; Hayakawa, T. Ganglioside GD1a inhibits HGF-induced motility and scattering of cancer cells through suppression of tyrosine phosphorylation of c-Met. Int. J. Cancer 2001, 94, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, R.K. Interaction of ganglioside GD3 with an EGF receptor sustains the self-renewal ability of mouse neural stem cells in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 19137–19142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, R.; Ladisch, S. Exogenous ganglioside GD1a enhances epidermal growth factor receptor binding and dimerization. J. Biol. Chem. 2004, 279, 36481–36489. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Jung, K.Y.; Kwak, D.H.; Lee, S.H.; Ryu, J.S.; Kim, J.S.; Chang, K.T.; Lee, J.W.; Choo, Y.K. Inhibition of ganglioside GD1a synthesis suppresses the differentiation of human mesenchymal stem cells into osteoblasts. Dev. Growth Differ. 2011, 53, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Horto-bagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.J.; Ding, Y.; Levery, S.B.; Lobaton, M.; Handa, K.; Hakomori, S.I. Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 4968–4973. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.R.; Battula, V.L.; Werden, S.J.; Vijay, G.V.; Ramirez-Peña, E.Q.; Taube, J.H.; Chang, J.T.; Miura, N.; Porter, W.; Sphyris, N.; et al. GD3 synthase regulates epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene 2015, 34, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
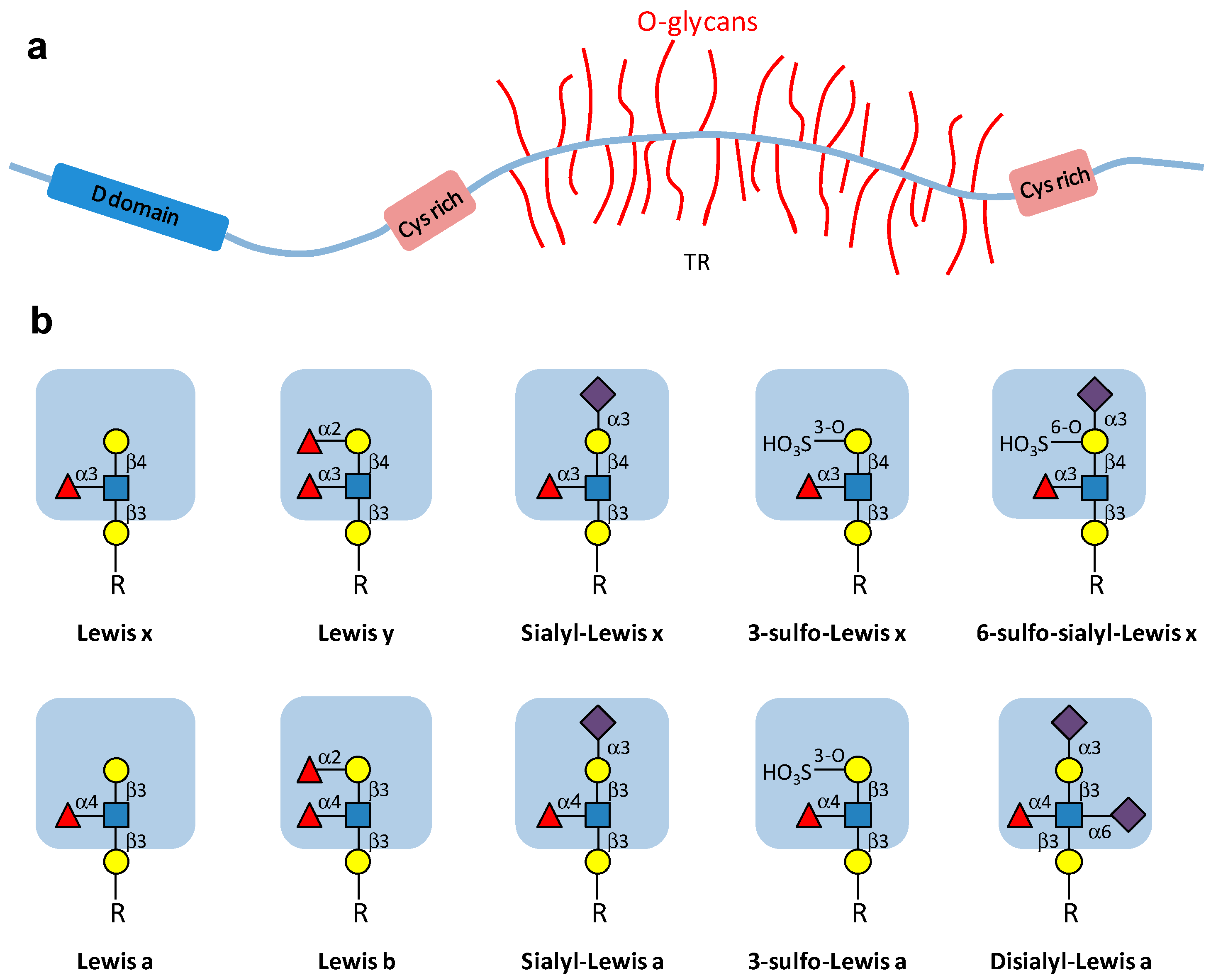
 : Gal;
: Gal;  : GlcNAc;
: GlcNAc;  : Fuc;
: Fuc;  : Neu5Ac; HO3S: sulfate. R: O-glycan chain.
: Gal; : GlcNAc; : Fuc; : Neu5Ac; HO3S: sulfate. R: O-glycan chain.
: Neu5Ac; HO3S: sulfate. R: O-glycan chain.
: Gal; : GlcNAc; : Fuc; : Neu5Ac; HO3S: sulfate. R: O-glycan chain.




© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dewald, J.H.; Colomb, F.; Bobowski-Gerard, M.; Groux-Degroote, S.; Delannoy, P. Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer. Cells 2016, 5, 43. https://doi.org/10.3390/cells5040043
Dewald JH, Colomb F, Bobowski-Gerard M, Groux-Degroote S, Delannoy P. Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer. Cells. 2016; 5(4):43. https://doi.org/10.3390/cells5040043
Chicago/Turabian StyleDewald, Justine H., Florent Colomb, Marie Bobowski-Gerard, Sophie Groux-Degroote, and Philippe Delannoy. 2016. "Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer" Cells 5, no. 4: 43. https://doi.org/10.3390/cells5040043
APA StyleDewald, J. H., Colomb, F., Bobowski-Gerard, M., Groux-Degroote, S., & Delannoy, P. (2016). Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer. Cells, 5(4), 43. https://doi.org/10.3390/cells5040043