Differential Location and Distribution of Hepatic Immune Cells

Abstract
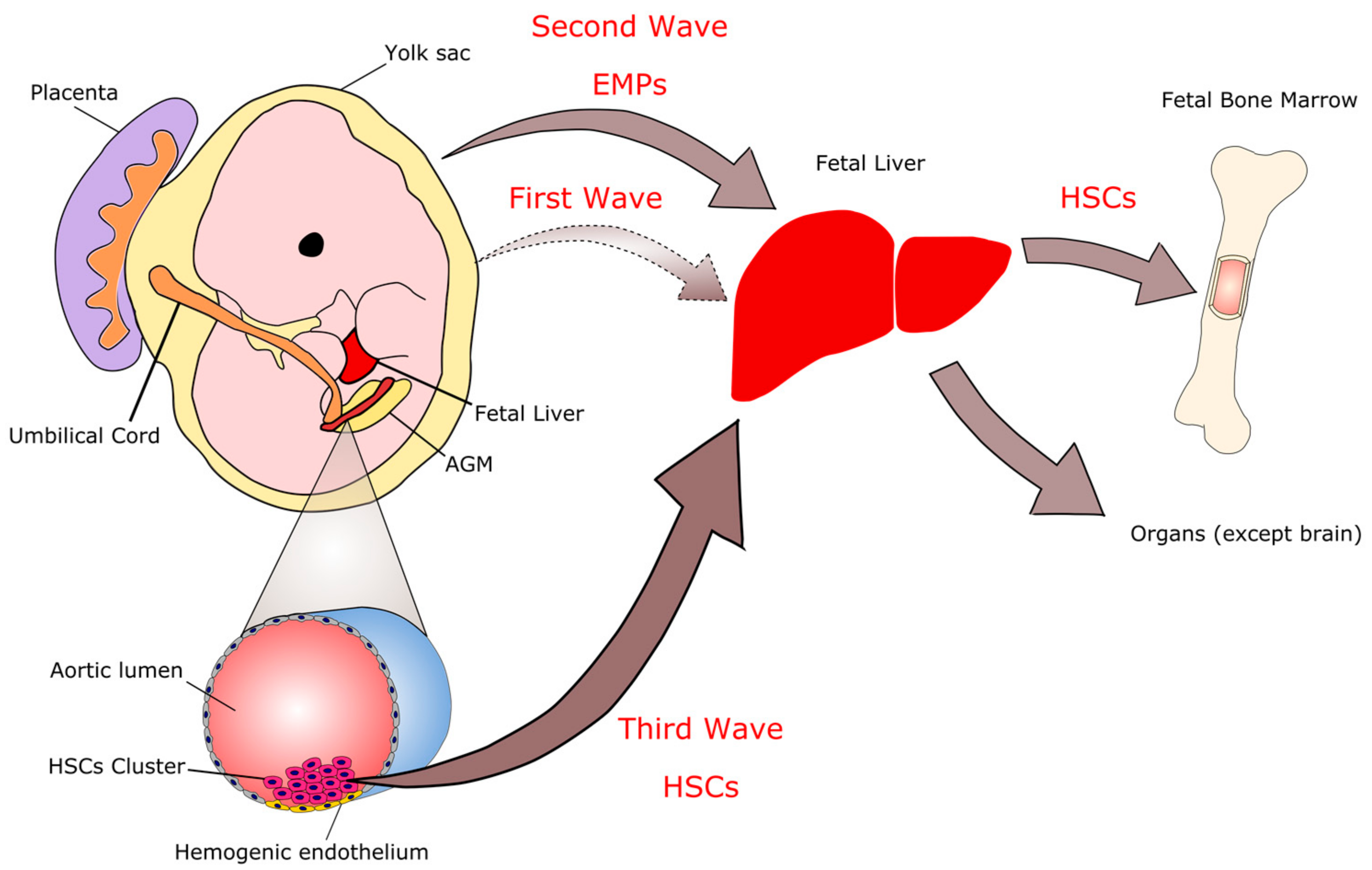
:1. Introduction
2. Immune System Ontogeny and the Correlation with the Fetal Liver
3. Differential Location of Immune Cells throughout the Liver
3.1. Phagocytes
3.1.1. Macrophages and Monocytes
3.1.2. Dendritic Cells
3.2. Granulocytes
3.2.1. Neutrophils
3.2.2. Eosinophils
3.3. Lymphocytes
3.3.1. Liver Natural Killer (NK)
3.3.2. Liver NKT Cells
3.3.3. Liver γδ T Cells
3.3.4. Liver B Cells
4. Concluding Remarks
Acknowledgments
Conflicts of Interest
Grant Support
References
- Molina, D.K.; DiMaio, V.J. Normal organ weights in men: Part ii-the brain, lungs, liver, spleen, and kidneys. Am. J. Forensic Med. Pathol. 2012, 33, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Eipel, C.; Abshagen, K.; Vollmar, B. Regulation of hepatic blood flow: The hepatic arterial buffer response revisited. World J. Gastroenterol. 2010, 16, 6046–6057. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tezuka, F. Hepatic arteries and arterial circulation in liver cirrhosis. Tohoku J. Exp. Med. 1974, 113, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.E.; Ingelfinger, F.J.; Bradley, G.P. Hepatic circulation in cirrhosis of the liver. Circulation 1952, 5, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Wisse, E.; Jacobs, F.; Topal, B.; Frederik, P.; De Geest, B. The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene Ther. 2008, 15, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Maslak, E.; Gregorius, A.; Chlopicki, S. Liver sinusoidal endothelial cells (lsecs) function and nafld; no-based therapy targeted to the liver. Pharmacol. Rep. 2015, 67, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Wake, K.; Decker, K.; Kirn, A.; Knook, D.L.; McCuskey, R.S.; Bouwens, L.; Wisse, E. Cell biology and kinetics of kupffer cells in the liver. Int. Rev. Cytol. 1989, 118, 173–229. [Google Scholar] [PubMed]
- Enzan, H.; Hara, H.; Yamashita, Y.; Ohkita, T.; Yamane, T. Fine structure of hepatic sinusoids and their development in human embryos and fetuses. Acta Pathol. Jpn. 1983, 33, 447–466. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamamura, F.; Naito, M. Differentiation, maturation, and proliferation of macrophages in the mouse yolk sac: A light-microscopic, enzyme-cytochemical, immunohistochemical, and ultrastructural study. J. Leukoc. Biol. 1989, 45, 87–96. [Google Scholar]
- Cline, M.J.; Moore, M.A. Embryonic origin of the mouse macrophage. Blood 1972, 39, 842–849. [Google Scholar] [PubMed]
- Fukuda, T. Fetal hemopoiesis. I. Electron microscopic studies on human yolk sac hemopoiesis. Virchows Arch. B Cell Pathol. 1973, 14, 197–213. [Google Scholar] [PubMed]
- Moore, M.A.; Metcalf, D. Ontogeny of the haemopoietic system: Yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br. J. Haematol. 1970, 18, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 1999, 126, 5073–5084. [Google Scholar] [PubMed]
- Tober, J.; Koniski, A.; McGrath, K.E.; Vemishetti, R.; Emerson, R.; de Mesy-Bentley, K.K.; Waugh, R.; Palis, J. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 2007, 109, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Takahashi, K.; Nishikawa, S. Development, differentiation, and maturation of macrophages in the fetal mouse liver. J. Leukoc. Biol. 1990, 48, 27–37. [Google Scholar] [PubMed]
- Bertrand, J.Y.; Jalil, A.; Klaine, M.; Jung, S.; Cumano, A.; Godin, I. Three pathways to mature macrophages in the early mouse yolk sac. Blood 2005, 106, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Hasegawa, G.; Takahashi, K. Development, differentiation, and maturation of kupffer cells. Microsc. Res. Tech. 1997, 39, 350–364. [Google Scholar] [CrossRef]
- Bertrand, J.Y.; Giroux, S.; Golub, R.; Klaine, M.; Jalil, A.; Boucontet, L.; Godin, I.; Cumano, A. Characterization of purified intraembryonic hematopoietic stem cells as a tool to define their site of origin. Proc. Natl. Acad. Sci. USA 2005, 102, 134–139. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.E.; Frame, J.M.; Fegan, K.H.; Bowen, J.R.; Conway, S.J.; Catherman, S.C.; Kingsley, P.D.; Koniski, A.D.; Palis, J. Distinct sources of hematopoietic progenitors emerge before hscs and provide functional blood cells in the mammalian embryo. Cell Rep. 2015, 11, 1892–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frame, J.M.; McGrath, K.E.; Palis, J. Erythro-myeloid progenitors: “Definitive” hematopoiesis in the conceptus prior to the emergence of hematopoietic stem cells. Blood Cells Mol. Dis 2013, 51, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Li, Y.; De Obaldia, M.E.; Yang, Q.; Yzaguirre, A.D.; Yamada-Inagawa, T.; Vink, C.S.; Bhandoola, A.; Dzierzak, E.; Speck, N.A. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell 2011, 9, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Yokomizo, T.; Zeigler, B.M.; Dzierzak, E.; Speck, N.A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 2009, 457, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Yoder, M.C. Yolk-sac hematopoiesis: The first blood cells of mouse and man. Exp. Hematol. 2001, 29, 927–936. [Google Scholar] [CrossRef]
- Kieusseian, A.; Brunet de la Grange, P.; Burlen-Defranoux, O.; Godin, I.; Cumano, A. Immature hematopoietic stem cells undergo maturation in the fetal liver. Development 2012, 139, 3521–3530. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via pu.1- and irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Sumner, R.; Crawford, A.; Mucenski, M.; Frampton, J. Initiation of adult myelopoiesis can occur in the absence of c-myb whereas subsequent development is strictly dependent on the transcription factor. Oncogene 2000, 19, 3335–3342. [Google Scholar] [CrossRef] [PubMed]
- Bartunek, P.; Kralova, J.; Blendinger, G.; Dvorak, M.; Zenke, M. Gata-1 and c-myb crosstalk during red blood cell differentiation through gata-1 binding sites in the c-myb promoter. Oncogene 2003, 22, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Godin, I.; Cumano, A. The hare and the tortoise: An embryonic haematopoietic race. Nat. Rev. Immunol. 2002, 2, 593–604. [Google Scholar] [PubMed]
- Zovein, A.C.; Turlo, K.A.; Ponec, R.M.; Lynch, M.R.; Chen, K.C.; Hofmann, J.J.; Cox, T.C.; Gasson, J.C.; Iruela-Arispe, M.L. Vascular remodeling of the vitelline artery initiates extravascular emergence of hematopoietic clusters. Blood 2010, 116, 3435–3444. [Google Scholar] [CrossRef] [PubMed]
- Delassus, S.; Cumano, A. Circulation of hematopoietic progenitors in the mouse embryo. Immunity 1996, 4, 97–106. [Google Scholar] [CrossRef]
- Mucenski, M.L.; McLain, K.; Kier, A.B.; Swerdlow, S.H.; Schreiner, C.M.; Miller, T.A.; Pietryga, D.W.; Scott, W.J., Jr.; Potter, S.S. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 1991, 65, 677–689. [Google Scholar] [CrossRef]
- Soza-Ried, C.; Hess, I.; Netuschil, N.; Schorpp, M.; Boehm, T. Essential role of c-myb in definitive hematopoiesis is evolutionarily conserved. Proc. Natl. Acad. Sci. USA 2010, 107, 17304–17308. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Chiba, S.; Kunisato, A.; Sata, M.; Saito, T.; Nakagami-Yamaguchi, E.; Yamaguchi, T.; Masuda, S.; Shimizu, K.; Takahashi, T.; et al. Notch1 but not notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity 2003, 18, 699–711. [Google Scholar] [CrossRef]
- Gregory, S.H.; Wing, E.J. Accessory function of kupffer cells in the antigen-specific blastogenic response of an l3t4+ t-lymphocyte clone to listeria monocytogenes. Infect. Immun. 1990, 58, 2313–2319. [Google Scholar] [PubMed]
- Bouwens, L.; Baekeland, M.; De Zanger, R.; Wisse, E. Quantitation, tissue distribution and proliferation kinetics of kupffer cells in normal rat liver. Hepatology 1986, 6, 718–722. [Google Scholar] [CrossRef] [PubMed]
- David, B.A.; Rezende, R.M.; Antunes, M.M.; Santos, M.M.; Freitas Lopes, M.A.; Diniz, A.B.; Sousa Pereira, R.V.; Marchesi, S.C.; Alvarenga, D.M.; Nakagaki, B.N.; et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology 2016, 151, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Benacerraf, B.; Sebestyen, M.M.; Schlossman, S. A quantitative study of the kinetics of blood clearance of p32-labelled escherichia coli and staphylococci by the reticuloendothelial system. J. Exp. Med. 1959, 110, 27–48. [Google Scholar] [CrossRef] [PubMed]
- MacPhee, P.J.; Schmidt, E.E.; Groom, A.C. Evidence for kupffer cell migration along liver sinusoids, from high-resolution in vivo microscopy. Am. J. Physiol. 1992, 263, G17–G23. [Google Scholar] [PubMed]
- Marques, P.E.; Oliveira, A.G.; Pereira, R.V.; David, B.A.; Gomides, L.F.; Saraiva, A.M.; Pires, D.A.; Novaes, J.T.; Patricio, D.O.; Cisalpino, D.; et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 2015, 61, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Sleyster, E.C.; Knook, D.L. Relation between localization and function of rat liver kupffer cells. Lab. Investig. 1982, 47, 484–490. [Google Scholar] [PubMed]
- Armbrust, T.; Ramadori, G. Functional characterization of two different kupffer cell populations of normal rat liver. J. Hepatol. 1996, 25, 518–528. [Google Scholar] [CrossRef]
- Kinoshita, M.; Uchida, T.; Sato, A.; Nakashima, M.; Nakashima, H.; Shono, S.; Habu, Y.; Miyazaki, H.; Hiroi, S.; Seki, S. Characterization of two f4/80-positive kupffer cell subsets by their function and phenotype in mice. J. Hepatol. 2010, 53, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chen, J.B.; Tsai, T.F.; Tsai, Y.C.; Tsai, C.Y.; Liang, P.H.; Hsu, T.L.; Wu, C.Y.; Netea, M.G.; Wong, C.H.; et al. Clec4f is an inducible c-type lectin in f4/80-positive cells and is involved in alpha-galactosylceramide presentation in liver. PLoS ONE 2013, 8, e65070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar] [PubMed]
- Yona, S.; Gordon, S. From the reticuloendothelial to mononuclear phagocyte system—The unaccounted years. Front. Immunol. 2015, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- Fogg, D.K.; Sibon, C.; Miled, C.; Jung, S.; Aucouturier, P.; Littman, D.R.; Cumano, A.; Geissmann, F. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 2006, 311, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.H.; Sathe, P.; Park, H.Y.; Metcalf, D.; Proietto, A.I.; Dakic, A.; Carotta, S.; O’Keeffe, M.; Bahlo, M.; Papenfuss, A.; et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 2007, 8, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Onai, N.; Obata-Onai, A.; Schmid, M.A.; Ohteki, T.; Jarrossay, D.; Manz, M.G. Identification of clonogenic common flt3+m-csfr+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat. Immunol. 2007, 8, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Victora, G.D.; Schwickert, T.A.; Guermonprez, P.; Meredith, M.M.; Yao, K.; Chu, F.F.; Randolph, G.J.; Rudensky, A.Y.; Nussenzweig, M. In vivo analysis of dendritic cell development and homeostasis. Science 2009, 324, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Liu, K.; Helft, J.; Bogunovic, M.; Greter, M.; Hashimoto, D.; Price, J.; Yin, N.; Bromberg, J.; Lira, S.A.; et al. The origin and development of nonlymphoid tissue cd103+ dcs. J. Exp. Med. 2009, 206, 3115–3130. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; van de Laar, L. A hitchhiker’s guide to myeloid cell subsets: Practical implementation of a novel mononuclear phagocyte classification system. Front. Immunol. 2015, 6, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, L.; Saelens, W.; De Prijck, S.; Martens, L.; Scott, C.L.; Van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.N.; et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Deimann, W.; Fahimi, H.D. Induction of focal hemopoiesis in adult rat liver by glucan, a macrophage activator. A cytochemical and ultrastructural study. Lab. Investig. 1980, 42, 217–224. [Google Scholar] [PubMed]
- Pino, R.M.; Bankston, P.W. The development of the sinusoids of fetal rat liver: Localization of endogenous peroxidase in fetal kupffer cells. J. Histochem. Cytochem. 1979, 27, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Wisse, E. Observations on the fine structure and peroxidase cytochemistry of normal rat liver kupffer cells. J. Ultrastruct. Res. 1974, 46, 393–426. [Google Scholar] [CrossRef]
- Wisse, E. Kupffer cell reactions in rat liver under various conditions as observed in the electron microscope. J. Ultrastruct. Res. 1974, 46, 499–520. [Google Scholar] [CrossRef]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varol, C.; Vallon-Eberhard, A.; Elinav, E.; Aychek, T.; Shapira, Y.; Luche, H.; Fehling, H.J.; Hardt, W.D.; Shakhar, G.; Jung, S. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity 2009, 31, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.I.; Tamaki, K.; Sachs, D.H. Epidermal langerhans cells are derived from cells originating in bone marrow. Nature 1979, 282, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Hickey, W.F.; Vass, K.; Lassmann, H. Bone marrow-derived elements in the central nervous system: An immunohistochemical and ultrastructural survey of rat chimeras. J. Neuropathol. Exp. Neurol. 1992, 51, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, C.; Dupuis, T.; Jouvion, G.; Eberl, G.; Disson, O.; Lecuit, M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 2015, 42, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Rosas, M.; Smith, P.J.; Fraser, D.J.; Jones, S.A.; Taylor, P.R. A quantifiable proliferative burst of tissue macrophages restores homeostatic macrophage populations after acute inflammation. Eur. J. Immunol. 2011, 41, 2155–2164. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Naito, M.; Takahashi, K. Kupffer cell proliferation and glucan-induced granuloma formation in mice depleted of blood monocytes by strontium-89. J. Leukoc. Biol. 1990, 47, 195–205. [Google Scholar] [PubMed]
- Bruttger, J.; Karram, K.; Wortge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 2015, 43, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Okabe, Y.; Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014, 157, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Helmy, K.Y.; Katschke, K.J., Jr.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. Crig: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006, 124, 915–927. [Google Scholar] [CrossRef] [PubMed]
- He, J.Q.; Katschke, K.J., Jr.; Gribling, P.; Suto, E.; Lee, W.P.; Diehl, L.; Eastham-Anderson, J.; Ponakala, A.; Komuves, L.; Egen, J.G.; et al. Crig mediates early kupffer cell responses to adenovirus. J. Leukoc. Biol. 2013, 93, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Broadley, S.P.; Plaumann, A.; Coletti, R.; Lehmann, C.; Wanisch, A.; Seidlmeier, A.; Esser, K.; Luo, S.; Ramer, P.C.; Massberg, S.; et al. Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe 2016, 20, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Surewaard, B.G.; Wong, C.H.; Geoghegan, J.A.; Jenne, C.N.; Kubes, P. Crig functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe 2016, 20, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Schurich, A.; Berg, M.; Stabenow, D.; Bottcher, J.; Kern, M.; Schild, H.J.; Kurts, C.; Schuette, V.; Burgdorf, S.; Diehl, L.; et al. Dynamic regulation of cd8 t cell tolerance induction by liver sinusoidal endothelial cells. J. Immunol. 2010, 184, 4107–4114. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.S.; Lu, B.; Sierro, F.; Benseler, V.; McGuffog, C.M.; Bishop, G.A.; Cowan, P.J.; McCaughan, G.W.; Dwyer, K.M.; Bowen, D.G.; et al. Differential migration of passenger leukocytes and rapid deletion of naive alloreactive cd8 t cells after mouse liver transplantation. Liver Transplant. 2013, 19, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Breous, E.; Somanathan, S.; Vandenberghe, L.H.; Wilson, J.M. Hepatic regulatory t cells and kupffer cells are crucial mediators of systemic t cell tolerance to antigens targeting murine liver. Hepatology 2009, 50, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Kruse, N.; Neumann, K.; Schrage, A.; Derkow, K.; Schott, E.; Erben, U.; Kuhl, A.; Loddenkemper, C.; Zeitz, M.; Hamann, A.; et al. Priming of cd4+ t cells by liver sinusoidal endothelial cells induces cd25low forkhead box protein 3- regulatory t cells suppressing autoimmune hepatitis. Hepatology 2009, 50, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by kupffer cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Schippers, M.; Reker-Smit, C.; Martinez, F.O.; Helming, L.; Poelstra, K.; Melgert, B.N. Hepatic localization of macrophage phenotypes during fibrogenesis and resolution of fibrosis in mice and humans. Front. Immunol. 2014, 5, 430. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 and interleukin-1 antagonism. Blood 1991, 77, 1627–1652. [Google Scholar] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 kupffer cells promote m1 kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Moriarty, T.J.; Wong, C.H.; Zhou, H.; Strieter, R.M.; van Rooijen, N.; Chaconas, G.; Kubes, P. An intravascular immune response to borrelia burgdorferi involves kupffer cells and inkt cells. Nat. Immunol. 2010, 11, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Elsegood, C.L.; Chan, C.W.; Degli-Esposti, M.A.; Wikstrom, M.E.; Domenichini, A.; Lazarus, K.; van Rooijen, N.; Ganss, R.; Olynyk, J.K.; Yeoh, G.C. Kupffer cell-monocyte communication is essential for initiating murine liver progenitor cell-mediated liver regeneration. Hepatology 2015, 62, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.X.; Okada, T.; He, X.S.; Kita, H.; Liu, Y.J.; Ansari, A.A.; Kikuchi, K.; Ikehara, S.; Gershwin, M.E. Heterogeneity of dendritic cells in the mouse liver: Identification and characterization of four distinct populations. J. Immunol. 2003, 170, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Steiniger, B.; Klempnauer, J.; Wonigeit, K. Phenotype and histological distribution of interstitial dendritic cells in the rat pancreas, liver, heart, and kidney. Transplantation 1984, 38, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.C.; McKenzie, J.L.; Hart, D.N. Characterization of interstitial dendritic cells in human liver. Transplantation 1988, 46, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Sierro, F.; Evrard, M.; Rizzetto, S.; Melino, M.; Mitchell, A.J.; Florido, M.; Beattie, L.; Walters, S.B.; Tay, S.S.; Lu, B.; et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity 2017, 47, 374–388.e376. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, R.L.; Shakhar, G.; Dudziak, D.; Wardemann, H.; Eisenreich, T.; Dustin, M.L.; Nussenzweig, M.C. Visualizing dendritic cell networks in vivo. Nat. Immunol. 2004, 5, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Drakes, M.L.; Zahorchak, A.F.; O’Connell, P.J.; Steptoe, R.J.; Qian, S.; Lu, L. Hepatic dendritic cells: Immunobiology and role in liver transplantation. J. Leukoc. Biol. 1999, 66, 322–330. [Google Scholar] [PubMed]
- Eckert, C.; Klein, N.; Kornek, M.; Lukacs-Kornek, V. The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front. Immunol. 2015, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Ardavin, C. Origin, precursors and differentiation of mouse dendritic cells. Nat. Rev. Immunol. 2003, 3, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Nussenzweig, M.C. Origin and development of dendritic cells. Immunol. Rev. 2010, 234, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Bunin, A.; Ghosh, H.S.; Lewis, K.L.; Sisirak, V. Plasmacytoid dendritic cells: Recent progress and open questions. Annu. Rev. Immunol. 2011, 29, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Boonstra, A.; Paturel, C.; Antonenko, S.; Xu, X.L.; Trinchieri, G.; O’Garra, A.; Liu, Y.J. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by flt3-ligand and granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 2002, 195, 953–958. [Google Scholar] [CrossRef] [PubMed]
- McGovern, N.; Shin, A.; Low, G.; Low, D.; Duan, K.; Yao, L.J.; Msallam, R.; Low, I.; Shadan, N.B.; Sumatoh, H.R.; et al. Human fetal dendritic cells promote prenatal t-cell immune suppression through arginase-2. Nature 2017, 546, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Pierre, P.; Turley, S.J.; Gatti, E.; Hull, M.; Meltzer, J.; Mirza, A.; Inaba, K.; Steinman, R.M.; Mellman, I. Developmental regulation of mhc class ii transport in mouse dendritic cells. Nature 1997, 388, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.R.; Graffeo, C.S.; Rehman, A.; Fallon, N.C.; Zambirinis, C.P.; Ochi, A.; Barilla, R.; Jamal, M.; Deutsch, M.; Greco, S.; et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013, 58, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, G.; Yang, H.R.; Gu, X.; Wang, L.; Hsieh, C.C.; Chou, H.S.; Fung, J.J.; Qian, S.; Lu, L. Distinct response of liver myeloid dendritic cells to endotoxin is mediated by il-27. J. Hepatol. 2009, 51, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Krueger, P.D.; Kim, T.S.; Sung, S.S.; Braciale, T.J.; Hahn, Y.S. Liver-resident cd103+ dendritic cells prime antiviral cd8+ t cells in situ. J. Immunol. 2015, 194, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Athens, J.W.; Haab, O.P.; Raab, S.O.; Mauer, A.M.; Ashenbrucker, H.; Cartwright, G.E.; Wintrobe, M.M. Leukokinetic studies. Iv. The total blood, circulating and marginal granulocyte pools and the granulocyte turnover rate in normal subjects. J. Clin Investig. 1961, 40, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, S.K.; Jaeschke, H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol. Pathol. 2007, 35, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Petri, B.; Phillipson, M.; Kubes, P. The physiology of leukocyte recruitment: An in vivo perspective. J. Immunol. 2008, 180, 6439–6446. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Hasegawa, T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006, 26, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Farhood, A.; McGuire, G.M.; Manning, A.M.; Miyasaka, M.; Smith, C.W.; Jaeschke, H. Intercellular adhesion molecule 1 (icam-1) expression and its role in neutrophil-induced ischemia-reperfusion injury in rat liver. J. Leukoc. Biol. 1995, 57, 368–374. [Google Scholar] [PubMed]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ho, Y.S.; Fisher, M.A.; Lawson, J.A.; Farhood, A. Glutathione peroxidase-deficient mice are more susceptible to neutrophil-mediated hepatic parenchymal cell injury during endotoxemia: Importance of an intracellular oxidant stress. Hepatology 1999, 29, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, M.; Arteel, G.E.; Smutney, O.M.; Conzelmann, L.O.; Zhong, Z.; Thurman, R.G.; Lemasters, J.J. Dependence of liver injury after hemorrhage/resuscitation in mice on nadph oxidase-derived superoxide. Shock 2003, 19, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gujral, J.S.; Hinson, J.A.; Jaeschke, H. Chlorotyrosine protein adducts are reliable biomarkers of neutrophil-induced cytotoxicity in vivo. Comp. Hepatol. 2004, 3 (Suppl. 1), S48. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y. Neutrophil myeloperoxidase and its substrates: Formation of specific markers and reactive compounds during inflammation. J. Clin. Biochem. Nutr. 2016, 58, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Weller, P.F. The immunobiology of eosinophils. N. Engl. J. Med. 1991, 324, 1110–1118. [Google Scholar] [PubMed]
- Rothenberg, M.E. Eosinophilia. N. Engl. J. Med. 1998, 338, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Dubucquoi, S.; Desreumaux, P.; Janin, A.; Klein, O.; Goldman, M.; Tavernier, J.; Capron, A.; Capron, M. Interleukin 5 synthesis by eosinophils: Association with granules and immunoglobulin-dependent secretion. J. Exp. Med. 1994, 179, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Pham, B.N.; Bemuau, J.; Durand, F.; Sauvanet, A.; Degott, C.; Prin, L.; Janin, A. Eotaxin expression and eosinophil infiltrate in the liver of patients with drug-induced liver disease. J. Hepatol. 2001, 34, 537–547. [Google Scholar] [CrossRef]
- Tarantino, G.; Cabibi, D.; Camma, C.; Alessi, N.; Donatelli, M.; Petta, S.; Craxi, A.; Di Marco, V. Liver eosinophilic infiltrate is a significant finding in patients with chronic hepatitis c. J. Viral Hepat. 2008, 15, 523–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornsson, E.; Kalaitzakis, E.; Olsson, R. The impact of eosinophilia and hepatic necrosis on prognosis in patients with drug-induced liver injury. Aliment. Pharmacol. Ther. 2007, 25, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D.G.; O’Farrelly, C. Innate and adaptive lymphoid cells in the human liver. Immunol. Rev. 2000, 174, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, J.P. Natural killer cell developmental pathways: A question of balance. Annu. Rev. Immunol. 2006, 24, 257–286. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, C.; Chasson, L.; Luci, C.; Tomasello, E.; Geissmann, F.; Vivier, E.; Walzer, T. The trafficking of natural killer cells. Immunol. Rev. 2007, 220, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Jiang, X.; Chen, Y.; Sojka, D.K.; Wei, H.; Gao, X.; Sun, R.; Yokoyama, W.M.; Tian, Z. Liver-resident nk cells confer adaptive immunity in skin-contact inflammation. J. Clin. Investig. 2013, 123, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Vermijlen, D.; Luo, D.; Froelich, C.J.; Medema, J.P.; Kummer, J.A.; Willems, E.; Braet, F.; Wisse, E. Hepatic natural killer cells exclusively kill splenic/blood natural killer-resistant tumor cells by the perforin/granzyme pathway. J. Leukoc. Biol. 2002, 72, 668–676. [Google Scholar] [PubMed]
- Ishiyama, K.; Ohdan, H.; Ohira, M.; Mitsuta, H.; Arihiro, K.; Asahara, T. Difference in cytotoxicity against hepatocellular carcinoma between liver and periphery natural killer cells in humans. Hepatology 2006, 43, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, S.I.; Thio, C.L.; Martin, M.P.; Brooks, C.R.; Gao, X.; Astemborski, J.; Cheng, J.; Goedert, J.J.; Vlahov, D.; Hilgartner, M.; et al. Hla and nk cell inhibitory receptor genes in resolving hepatitis c virus infection. Science 2004, 305, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, H.; Gao, B.; Hu, Z.; Zheng, S.; Tian, Z. Activation and function of hepatic nk cells in hepatitis b infection: An underinvestigated innate immune response. J. Viral Hepat. 2005, 12, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Subleski, J.J.; Hall, V.L.; Back, T.C.; Ortaldo, J.R.; Wiltrout, R.H. Enhanced antitumor response by divergent modulation of natural killer and natural killer t cells in the liver. Cancer Res. 2006, 66, 11005–11012. [Google Scholar] [CrossRef] [PubMed]
- Wisse, E.; van’t Noordende, J.M.; van der Meulen, J.; Daems, W.T. The pit cell: Description of a new type of cell occurring in rat liver sinusoids and peripheral blood. Cell Tissue Res. 1976, 173, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell. Mol. Immunol. 2016, 13, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, N.; Beziat, V.; Nystrom, S.; Hengst, J.; Ivarsson, M.A.; Kekalainen, E.; Johansson, H.; Mjosberg, J.; Westgren, M.; Lankisch, T.O.; et al. Cutting edge: Identification and characterization of human intrahepatic cd49a+ nk cells. J. Immunol. 2015, 194, 2467–2471. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wei, H.; Sun, R.; Tian, Z. The roles of innate immune cells in liver injury and regeneration. Cell. Mol. Immunol. 2007, 4, 241–252. [Google Scholar] [PubMed]
- Geissmann, F.; Cameron, T.O.; Sidobre, S.; Manlongat, N.; Kronenberg, M.; Briskin, M.J.; Dustin, M.L.; Littman, D.R. Intravascular immune surveillance by cxcr6+ nkt cells patrolling liver sinusoids. PLoS Biol. 2005, 3, e113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, R.M.; Lin, M.Y.; Lohman, B.L.; Varga, S.M.; Zarozinski, C.C.; Selin, L.K. Alpha beta and gamma delta t-cell networks and their roles in natural resistance to viral infections. Immunol. Rev. 1997, 159, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Role of gamma-delta t cells in liver inflammation and fibrosis. World J. Gastrointest. Pathophysiol. 2014, 5, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Lalor, P.F.; Shields, P.; Grant, A.; Adams, D.H. Recruitment of lymphocytes to the human liver. Immunol. Cell Biol. 2002, 80, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Probert, C.S.; Christ, A.D.; Saubermann, L.J.; Turner, J.R.; Chott, A.; Carr-Locke, D.; Balk, S.P.; Blumberg, R.S. Analysis of human common bile duct-associated t cells: Evidence for oligoclonality, t cell clonal persistence, and epithelial cell recognition. J. Immunol. 1997, 158, 1941–1948. [Google Scholar] [PubMed]
- Racanelli, V.; Sansonno, D.; Piccoli, C.; D’Amore, F.P.; Tucci, F.A.; Dammacco, F. Molecular characterization of b cell clonal expansions in the liver of chronically hepatitis c virus-infected patients. J. Immunol. 2001, 167, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.; Shimizu, Y.; Kashii, Y.; Kato, T.; Minemura, M.; Okada, K.; Nambu, S.; Takahara, T.; Higuchi, K.; Maeda, Y.; et al. Functional b-cell response in intrahepatic lymphoid follicles in chronic hepatitis c. Hepatology 1999, 30, 143–150. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liver Immune Cells | Location in Homeostasis | Main Surface Markers | Putative Role in Diseases |
|---|---|---|---|
| Kupffer Cells | Inside the sinusoids; adhered to the endothelium | F4/80, CD11b | Controlling inflammation; Kupffer cell depletion is associated with worse prognosis |
| Dendritic Cells | Underneath hepatic capsule; around large vessels | CD19−CD11c+; CD8α+B220− CD11b− (lymphoid); CD8α− B220−CD11b+ (myeloid); B220+ CD11b− (plasmacytoid) | Enhanced response to viral infections, controlling viral spread and T cell activation |
| Monocytes | Inside the sinusoids as patrolling cells | CD11bhiCD115hiGr1lo | Infiltrating monocytes control pathogen spread and heal tissue injury |
| Neutrophils | Ly6G+CD11b+F4/80− | Overt infiltration is associated with enhanced liver injury in several models | |
| Eosinophils | CD11b+CD193+Siglec F+ | Role in pathogenesis of liver diseases through release of granules containing TNF-α, highly cytotoxic proteins such as major basic protein and eosinophilic cationic protein | |
| Natural Killer Cells | CD3−NK1.1+ | Involved in the pathogenesis of liver diseases, mainly tumors and viral infections; higher cytotoxicity than other NK cells | |
| NKT Cells | CD3+NK1.1+ | Patrolling liver sinusoids to provide intravascular immune surveillance | |
| T lymphocytes | CD3+CD4+ (T CD4 cells); CD3+CD8+ (T CD8 cells) | Clearance of virus and in virus-induced immunopathology | |
| B lymphocytes | CD19+ | Antibody-secreting cells within germinal centers of intraportal lymphoid follicles, during viral hepatitis | |
| γδ T cells | CD24+ CD25− CD27+ | Controlling early viral infections; expressing perforin, lysing virus-infected targets, and releasing IFN-γ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitas-Lopes, M.A.; Mafra, K.; David, B.A.; Carvalho-Gontijo, R.; Menezes, G.B. Differential Location and Distribution of Hepatic Immune Cells. Cells 2017, 6, 48. https://doi.org/10.3390/cells6040048
Freitas-Lopes MA, Mafra K, David BA, Carvalho-Gontijo R, Menezes GB. Differential Location and Distribution of Hepatic Immune Cells. Cells. 2017; 6(4):48. https://doi.org/10.3390/cells6040048
Chicago/Turabian StyleFreitas-Lopes, Maria Alice, Kassiana Mafra, Bruna A. David, Raquel Carvalho-Gontijo, and Gustavo B. Menezes. 2017. "Differential Location and Distribution of Hepatic Immune Cells" Cells 6, no. 4: 48. https://doi.org/10.3390/cells6040048
APA StyleFreitas-Lopes, M. A., Mafra, K., David, B. A., Carvalho-Gontijo, R., & Menezes, G. B. (2017). Differential Location and Distribution of Hepatic Immune Cells. Cells, 6(4), 48. https://doi.org/10.3390/cells6040048