Perturbations in Traffic: Aberrant Nucleocytoplasmic Transport at the Heart of Neurodegeneration


Abstract
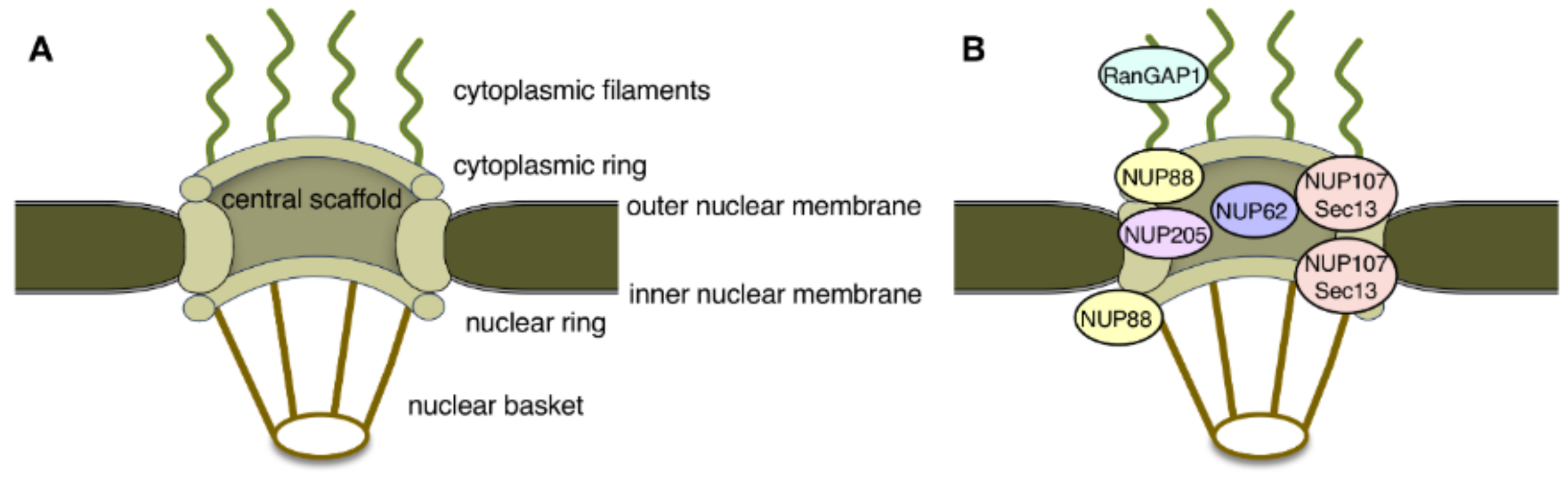
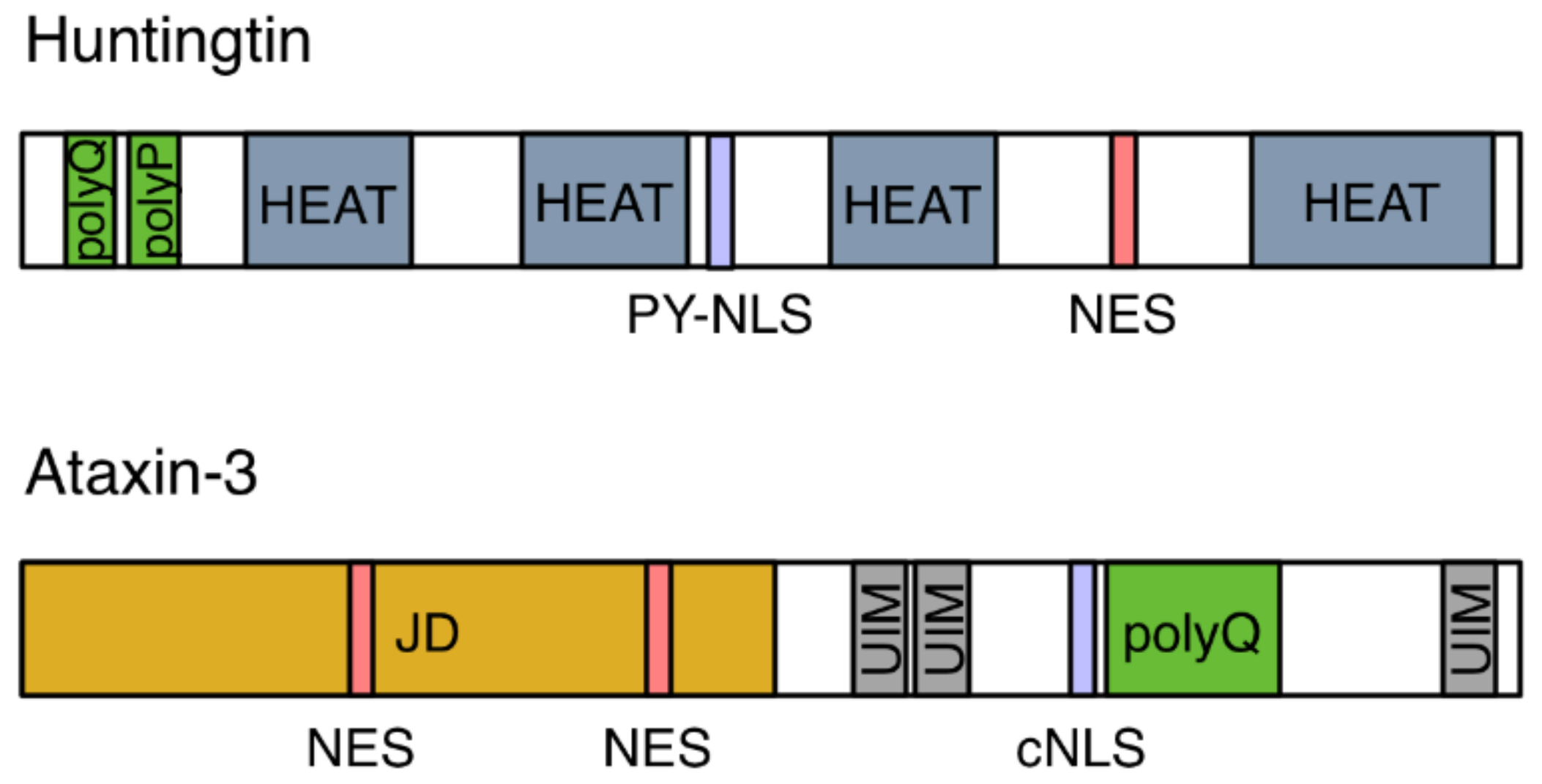
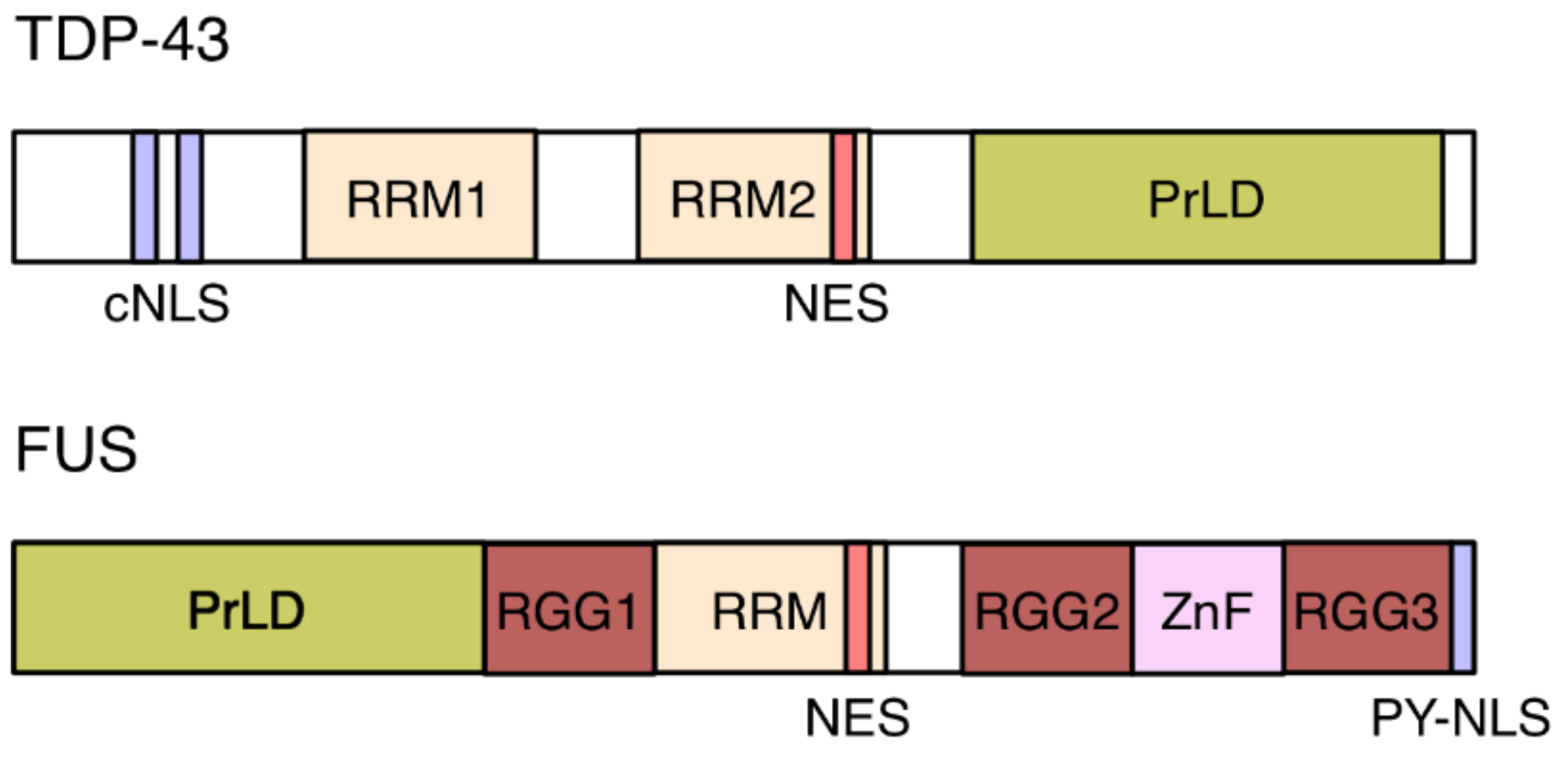
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
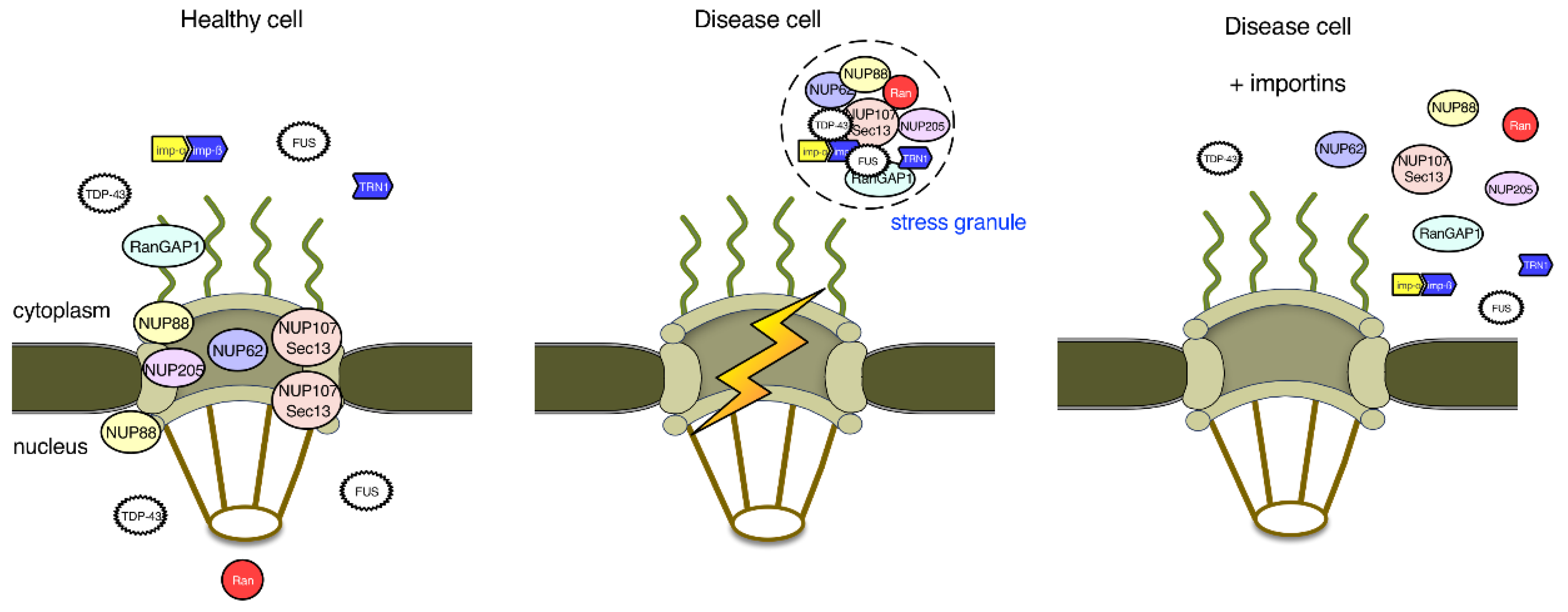{kind=link}
1. Defects Point to Nuclear Pores
2. Nucleocytoplasmic Transport of RNA-Binding Proteins
3. Chaperone Activity of Nuclear Import Receptors
4. Conclusions
Funding
Conflicts of Interest
References
- Zhang, K.; Grima, J.C.; Rothstein, J.D.; Lloyd, T.E. Nucleocytoplasmic transport in C9orf72-mediated ALS/FTD. Nucleus 2016, 7, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldman, M.B.; Yang, X.W. Huntington’s disease: Nuclear gatekeepers under attack. Neuron 2017, 94, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hezwani, M.; Fahrenkrog, B. The functional versatility of the nuclear pore complex proteins. Semin. Cell Dev. Biol. 2017, 68, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Fried, H.; Kutay, U. Nucleocytoplasmic transport: Taking an inventory. Cell Mol. Life Sci. 2003, 60, 1659–1688. [Google Scholar] [CrossRef] [PubMed]
- Bessert, D.A.; Gutridge, K.L.; Dunbar, J.C.; Carlock, L.R. The identification of a functional nuclear localization signal in the huntington disease protein. Mol. Brain Res. 1995, 33, 165–173. [Google Scholar] [CrossRef]
- Desmond, C.R.; Atwal, R.S.; Xia, J.; Truant, R. Identification of a karyopherin beta1/beta2 proline-tyrosine nuclear localization signal in huntingtin protein. J. Biol. Chem. 2012, 287, 39626–39633. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Lee, D.H.; Taylor, J.; Vandelft, M.; Truant, R. Huntingtin contains a highly conserved nuclear export signal. Hum. Mol. Genet. 2003, 12, 1393–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhr, S.T.; Senut, M.C.; Whitelegge, J.P.; Faull, K.F.; Cuizon, D.B.; Gage, F.H. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 2001, 153, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Hosp, F.; Vossfeldt, H.; Heinig, M.; Vasiljevic, D.; Arumughan, A.; Wyler, E.; The Genetic and Environmental Risk for Alzheimer’s Disease Consortium; Landthaler, M.; Hubner, N.; et al. Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 2015, 11, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.C.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron 2017, 94, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Bui, K.H.; von Appen, A.; DiGuilio, A.L.; Ori, A.; Sparks, L.; Mackmull, M.T.; Bock, T.; Hagen, W.; Andres-Pons, A.; Glavy, J.S.; et al. Integrated structural analysis of the human nuclear pore complex scaffold. Cell 2013, 155, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Lussi, Y.C.; Hugi, I.; Laurell, E.; Kutay, U.; Fahrenkrog, B. The nucleoporin Nup88 is interacting with nuclear lamina. Mol. Biol. Cell 2011, 22, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Gasset-Rosa, F.; Chillon-Marinas, C.; Goginashvili, A.; Atwal, R.S.; Artates, J.W.; Tabet, R.; Wheeler, V.C.; Bang, A.G.; Cleveland, D.W.; Lagier-Tourenne, C. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 2017, 94, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Ruba, A.; Yang, W. O-Glcnac-ylation in the nuclear pore complex. Cell Mol. Bioeng. 2016, 9, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Kohler, J.J. Glycosylation of the nuclear pore. Traffic 2014, 15, 347–361. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9ORF72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Yamashita, T.; Nakano, Y.; Morihara, R.; Li, X.; Feng, T.; Liu, X.; Huang, Y.; Fukui, Y.; Hishikawa, N.; et al. Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience 2017, 350, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Ito, H.; Hirano, A.; Fujita, K.; Wate, R.; Nakamura, M.; Kaneko, S.; Nakano, S.; Kusaka, H. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Battistini, S.; Ricci, C.; Lotti, E.M.; Benigni, M.; Gagliardi, S.; Zucco, R.; Bondavalli, M.; Marcello, N.; Ceroni, M.; Cereda, C. Severe familial ALS with a novel exon 4 mutation (L106F) in the SOD1 gene. J. Neurol. Sci. 2010, 293, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell 2018, 173, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of als pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in c9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Steyaert, J.; Scheveneels, W.; Vanneste, J.; Van Damme, P.; Robberecht, W.; Callaerts, P.; Bogaert, E.; Van Den Bosch, L. FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum. Mol. Genet. 2018, 27, 4103–4116. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Bogaert, E.; Van Damme, P.; Van Den Bosch, L. Inside out: The role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016, 132, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Taylor, J.P. Lost in transportation: Nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.; Neumann, M.; et al. Arginine methylation next to the PY-NLS modulates transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Zupunski, V.; Troakes, C.; Kathe, C.; Fratta, P.; Howell, M.; Gallo, J.M.; Hortobagyi, T.; Shaw, C.E.; Rogelj, B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 2010, 133, 1763–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Disturbance of nuclear and cytoplasmic tar DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovicic, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., 3rd; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.; Mantele, S.; Mollenkopf, P.; Boy, J.; Kehlenbach, R.H.; Riess, O.; Schmidt, T. Identification and functional dissection of localization signals within ataxin-3. Neurobiol. Dis. 2009, 36, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Sowa, A.S.; Martin, E.; Martins, I.M.; Schmidt, J.; Depping, R.; Weber, J.J.; Rother, F.; Hartmann, E.; Bader, M.; Riess, O.; et al. Karyopherin α-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3. Proc. Natl. Acad. Sci. USA 2018, 115, E2624–E2633. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Funk, C.; Abou-Ajram, C.; Hutten, S.; Funk, E.B.E.; Kehlenbach, R.H.; Bailer, S.M.; Dormann, D. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci. Rep. 2018, 8, 7084. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein phase separation: A new phase in cell biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Weber, C.A.; Julicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S. Phase separation in biology. Curr. Biol. 2017, 27, R1097–R1102. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhaleva, S.; Lemke, E.A. Beyond the transport function of import receptors: What’s all the fus about? Cell 2018, 173, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase separation of fus is suppressed by its nuclear import receptor and arginine methylation. Cell 2018, 173, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Strohl, F.; et al. Fus phase separation is modulated by a molecular chaperone and methylation of arginine cation-pi interactions. Cell 2018, 173, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear import receptor inhibits phase separation of fus through binding to multiple sites. Cell 2018, 173, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-import receptors reverse aberrant phase transitions of rna-binding proteins with prion-like domains. Cell 2018, 173, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Jakel, S.; Mingot, J.M.; Schwarzmaier, P.; Hartmann, E.; Gorlich, D. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J. 2002, 21, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, H.J. Prion-like mechanism in amyotrophic lateral sclerosis: Are protein aggregates the key? Exp. Neurobiol. 2015, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Shimonaka, S.; Nonaka, T.; Suzuki, G.; Hisanaga, S.; Hasegawa, M. Templated aggregation of tar DNA-binding protein of 43 kDa (TDP-43) by seeding with TDP-43 peptide fibrils. J. Biol. Chem. 2016, 291, 8896–8907. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 4220. [Google Scholar] [CrossRef] [PubMed]
- Svahn, A.J.; Don, E.K.; Badrock, A.P.; Cole, N.J.; Graeber, M.B.; Yerbury, J.J.; Chung, R.; Morsch, M. Nucleo-cytoplasmic transport of TDP-43 studied in real time: Impaired microglia function leads to axonal spreading of TDP-43 in degenerating motor neurons. Acta Neuropathol. 2018, 136, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau protein disrupts nucleocytoplasmic transport in alzheimer’s disease. Neuron 2018, 99, 925–940. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yang, J.; Choe, Y.J.; Hao, X.; Cao, X.; Zhao, Q.; Zhang, Y.; Franssens, V.; Hartl, F.U.; Nystrom, T.; et al. Role of the ribosomal quality control machinery in nucleocytoplasmic translocation of polyQ-expanded huntingtin exon-1. Biochem. Biophys. Res. Commun. 2017, 493, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.R.; Lundberg, L.; Pinkham, C.; Shechter, S.; DeBono, A.; Baell, J.; Wagstaff, K.M.; Hick, C.A.; Kehn-Hall, K.; Jans, D.A. Identification of novel antivirals inhibiting recognition of venezuelan equine encephalitis virus capsid protein by the importin alpha/beta1 heterodimer through high-throughput screening. Antiviral. Res. 2018, 151, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Cagatay, T.; Chook, Y.M. Karyopherins in cancer. Curr. Opin. Cell Biol. 2018, 52, 30–42. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahrenkrog, B.; Harel, A. Perturbations in Traffic: Aberrant Nucleocytoplasmic Transport at the Heart of Neurodegeneration. Cells 2018, 7, 232. https://doi.org/10.3390/cells7120232
Fahrenkrog B, Harel A. Perturbations in Traffic: Aberrant Nucleocytoplasmic Transport at the Heart of Neurodegeneration. Cells. 2018; 7(12):232. https://doi.org/10.3390/cells7120232
Chicago/Turabian StyleFahrenkrog, Birthe, and Amnon Harel. 2018. "Perturbations in Traffic: Aberrant Nucleocytoplasmic Transport at the Heart of Neurodegeneration" Cells 7, no. 12: 232. https://doi.org/10.3390/cells7120232
APA StyleFahrenkrog, B., & Harel, A. (2018). Perturbations in Traffic: Aberrant Nucleocytoplasmic Transport at the Heart of Neurodegeneration. Cells, 7(12), 232. https://doi.org/10.3390/cells7120232