PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Ethical Statement
2.4. Cell Proliferation Assays
2.5. Flow Cytometric Analysis of Cell Cycle Phase Distribution
2.6. p-Histone H3 Staining
2.7. Annexin-V Staining
2.8. Chromatin Fragmentation
2.9. Western Blot Analysis
2.10. Immunoprecipitation
2.11. Apoptosis Array
2.12. Statistical Analysis
3. Results
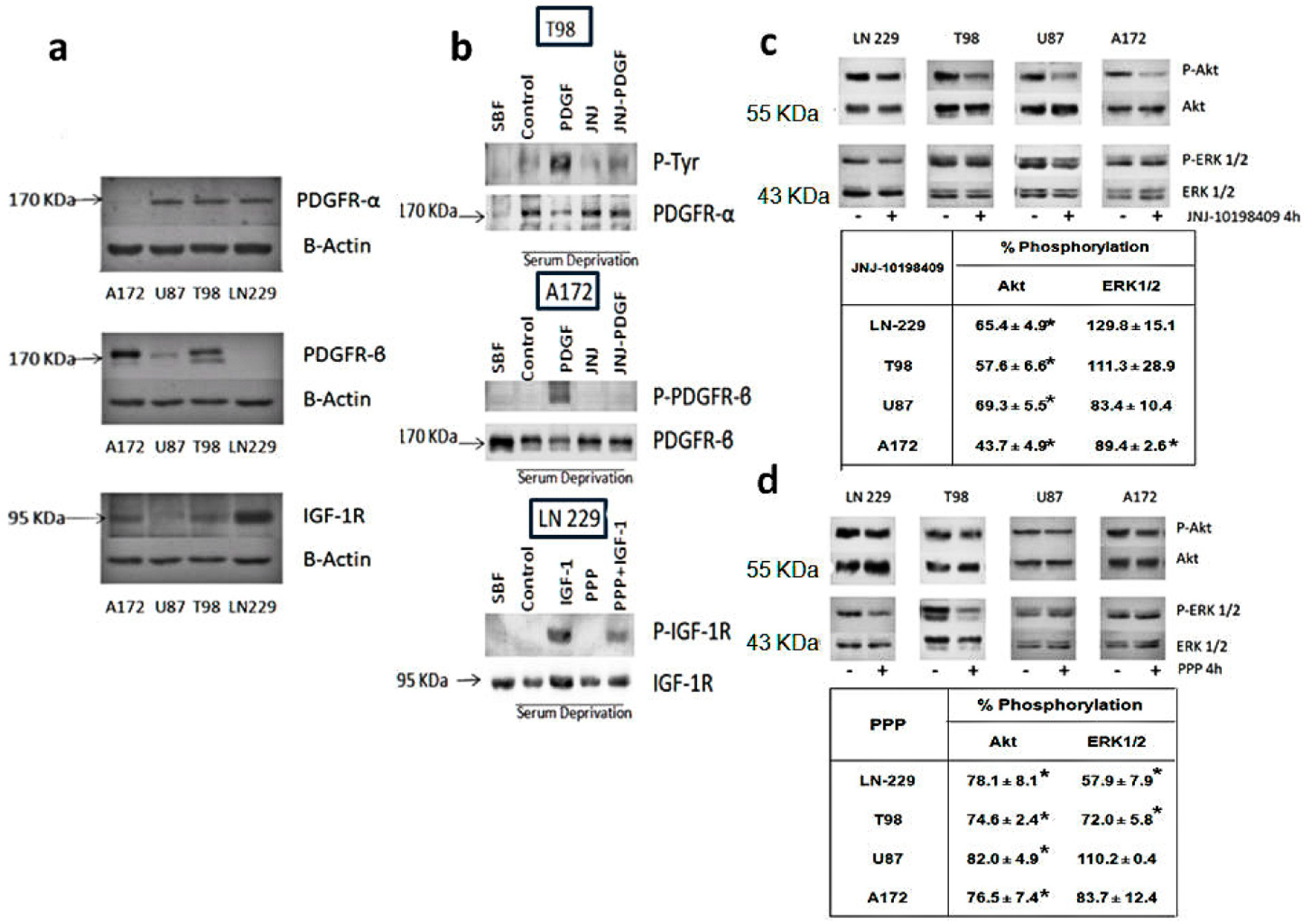
3.1. Expression Levels of PDGFR and IGF-1R in Glioblastoma Cell Lines
3.2. Inhibition of Phosphorylation of PDGFR, IGFR-1R and Downstream Pathways
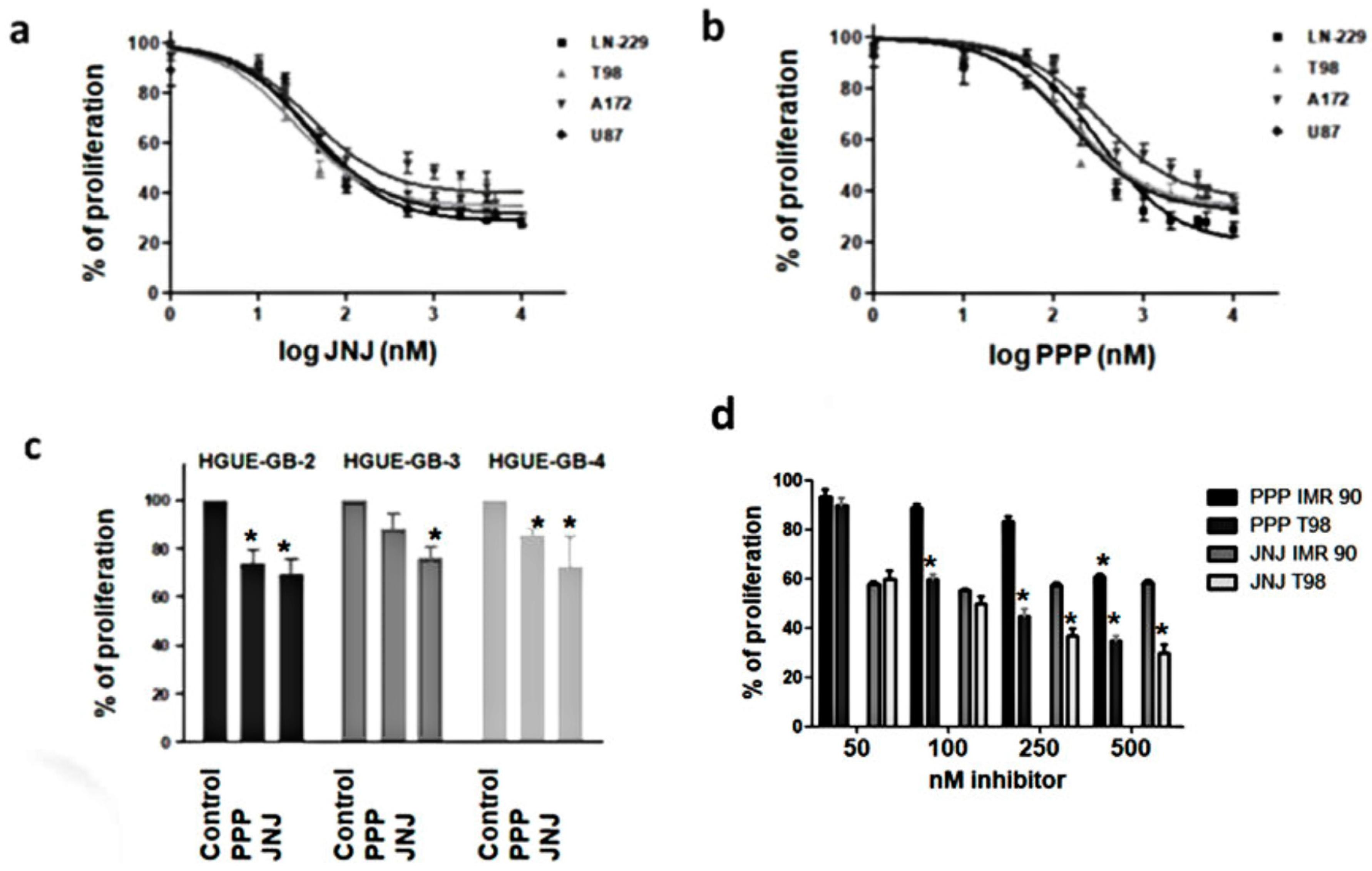
3.3. JNJ and PPP Inhibit Proliferation of Glioblastoma Cell Lines and Primary Cultures
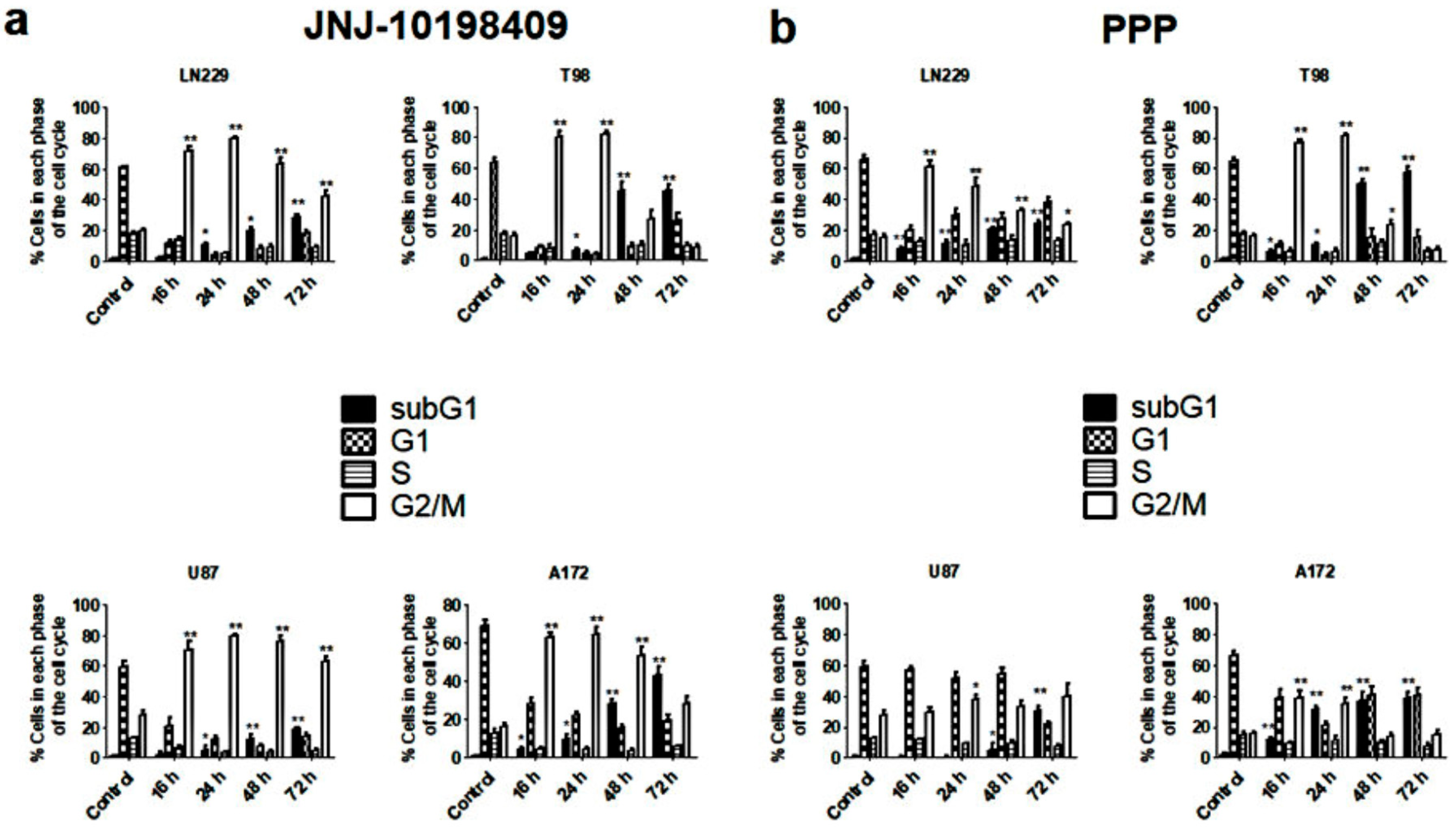
3.4. Effects of JNJ and PPP on Glioblastoma Cell Cycle
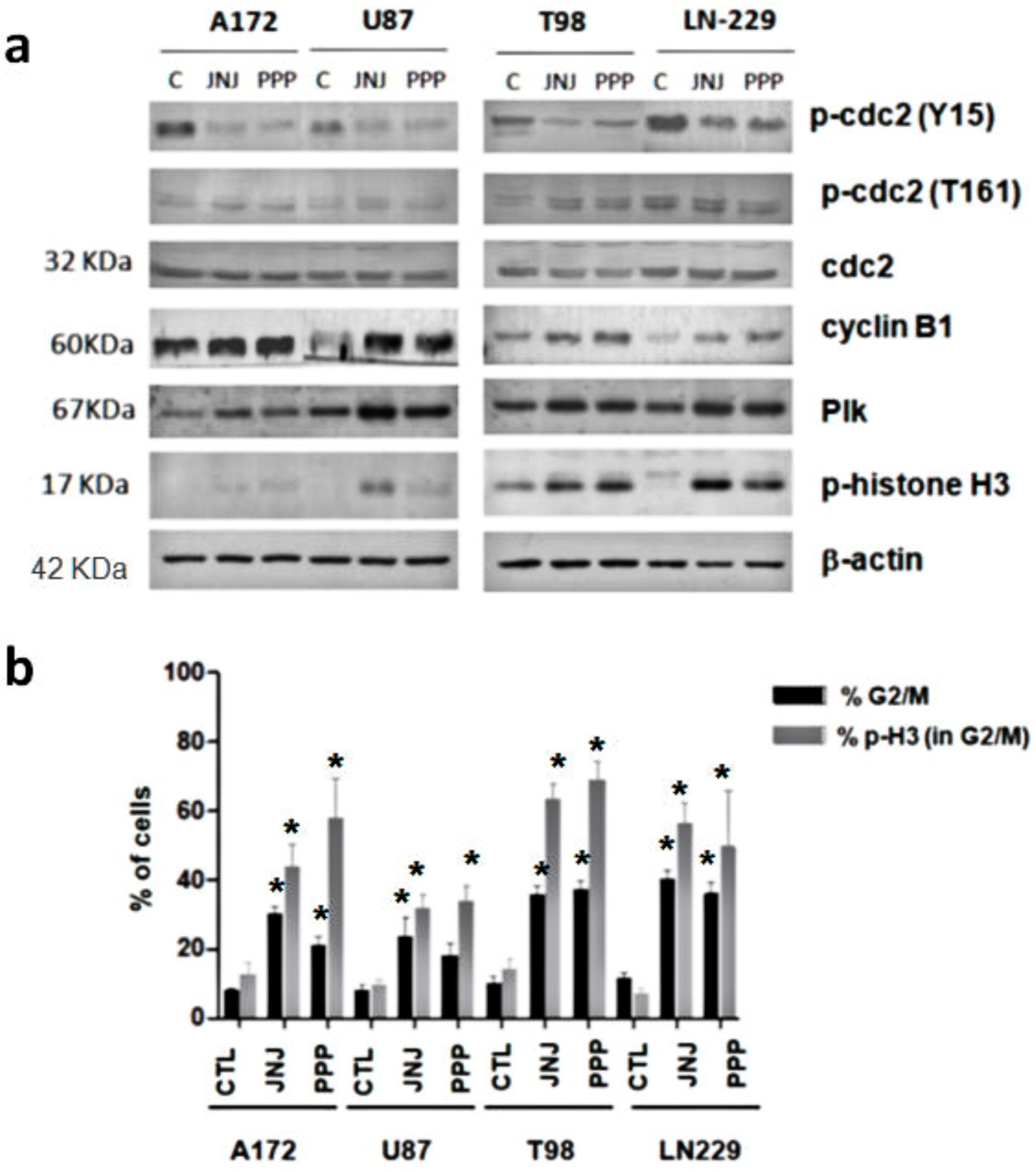
3.5. G2/M Arrest Induced by JNJ and PPP
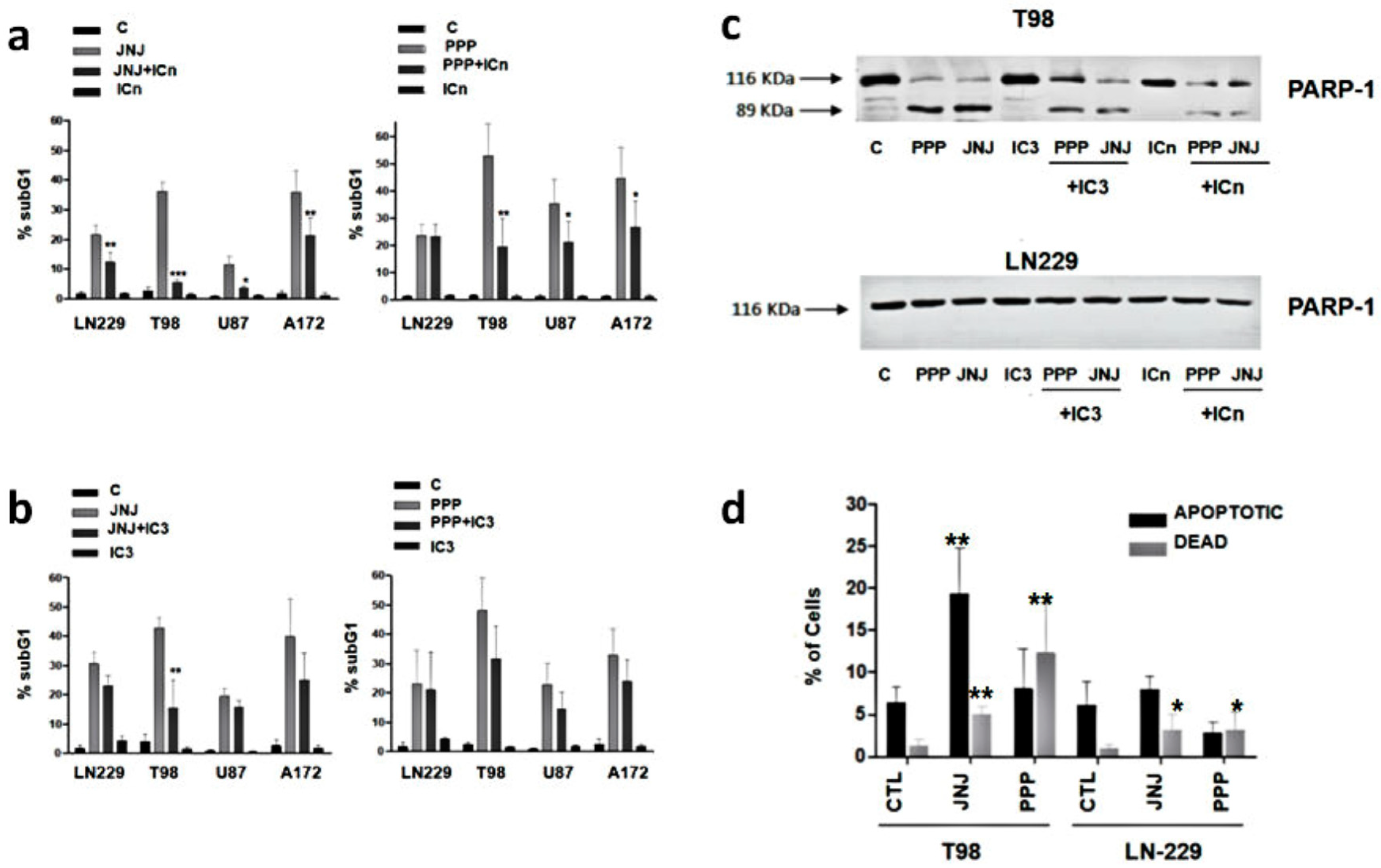
3.6. Are Caspases Involved in JNJ- or PPP-Induced Programmed Cell Death?
3.7. Is Apoptotic Cell Death Induced by JNJ or PPP?
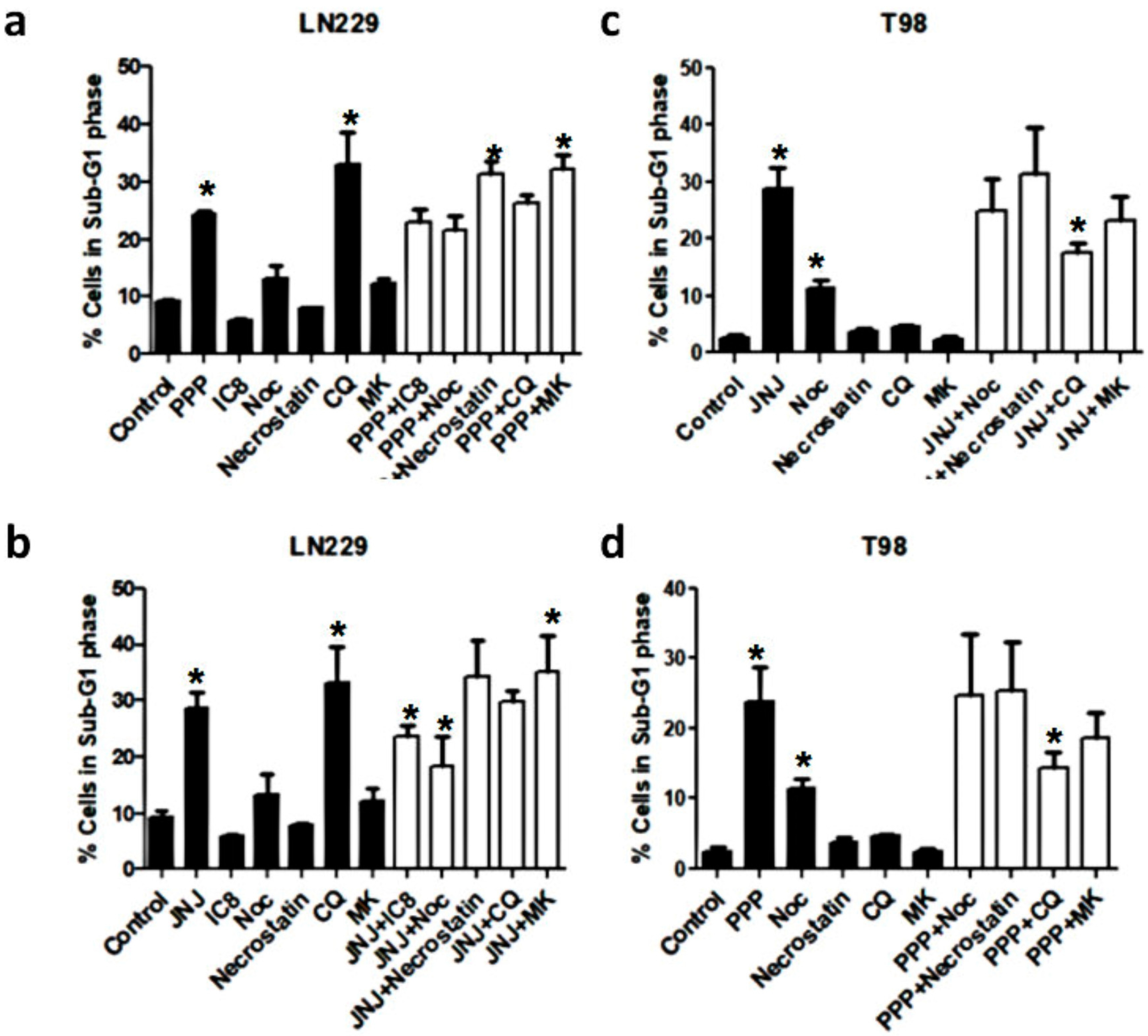
3.8. Alternative Mechanisms of Programmed Cell Death
3.9. Molecular Characterization of JNJ- and PPP-Induced Cell Death
3.10. Combination of PDGFR and IGF-1R Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gurney, J.G.; Kadan-Lottick, N. Brain and other central nervous system tumors: Rates, trends, and epidemiology. Curr. Opin. Oncol. 2001, 13, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Van Den Bent, M.J.; Mason, W.P.; Weller, M.; Mirimanoff, R.O.; Cairncross, J.G. Changing paradigms—An update on the multidisciplinary management of malignant glioma. Oncologist 2006, 11, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Villano, J.L.; Seery, T.E.; Bressler, L.R. Temozolomide in malignant gliomas: Current use and future targets. Cancer Chemother. Pharmacol. 2009, 64, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, M.; Funa, K.; Koopmann, J.; Maintz, D.; Waha, A.; Westermark, B.; Heldin, C.H.; Wiestler, O.D.; Louis, D.N.; Von Deimling, A.; et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor α receptor expression in human malignant gliomas. Cancer Res. 1996, 56, 164–171. [Google Scholar] [PubMed]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Garcia, E.; Saceda, M.; Martinez-Lacaci, I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells 2014, 3, 199–235. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Clinical.Trial.gov. Available online: http://www.clinicaltrials.gov/ct2/results?term=PDGFR%2C+glioma&Search=Search.
- Ho, C.Y.; Ludovici, D.W.; Maharoof, U.S.; Mei, J.; Sechler, J.L.; Tuman, R.W.; Strobel, E.D.; Andraka, L.; Yen, H.K.; Leo, G.; et al. (6,7-Dimethoxy-2,4-dihydroindeno[1,2-c]pyrazol-3-yl)phenylamines: Platelet-derived growth factor receptor tyrosine kinase inhibitors with broad antiproliferative activity against tumor cells. J. Med. Chem. 2005, 48, 8163–8173. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.R.; Mei, J.M.; Tuman, R.W.; Galemmo, R.A.; Johnson, D.L. Validation of in vivo pharmacodynamic activity of a novel PDGF receptor tyrosine kinase inhibitor using immunohistochemistry and quantitative image analysis. Mol. Cancer Ther. 2005, 4, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resnicoff, M.; Sell, C.; Rubini, M.; Coppola, D.; Ambrose, D.; Baserga, R.; Rubin, R. Rat glioblastoma cells expressing an antisense RNA to the insulin-like growth factor-1 (IGF-1) receptor are nontumorigenic and induce regression of wild-type tumors. Cancer Res. 1994, 54, 2218–2222. [Google Scholar] [PubMed]
- Resnicoff, M.; Li, W.; Basak, S.; Herlyn, D.; Baserga, R.; Rubin, R. Inhibition of rat C6 glioblastoma tumor growth by expression of insulin-like growth factor I receptor antisense mRNA. Cancer Immunol. Immunother. 1996, 42, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Rininsland, F.; Johnson, T.R.; Chernicky, C.L.; Schulze, E.; Burfeind, P.; Ilan, J. Suppression of insulin-like growth factor type I receptor by a triple-helix strategy inhibits IGF-I transcription and tumorigenic potential of rat C6 glioblastoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 5854–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girnita, A.; Girnita, L.; Del Prete, F.; Bartolazzi, A.; Larsson, O.; Axelson, M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 2004, 64, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [Google Scholar] [PubMed]
- Yin, S.; Girnita, A.; Strömberg, T.; Khan, Z.; Andersson, S.; Zheng, H.; Ericsson, C.; Axelson, M.; Nistér, M.; Larsson, O.; et al. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro. Oncol. 2009, 12, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin. Cancer Res. 2012, 18, 40–50. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Clinical.Trial.gov. Available online: http://www.clinicaltrials.gov/ct2/results?term=IGF1R%2C+glioma&Search=Search.
- Petronczki, M.; Lenart, P.; Peters, J.M. Polo on the Rise—From Mitotic Entry to Cytokinesis with Plk1. Dev. Cell 2008, 14, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Mizzen, C.A.; Cook, R.G.; Gorovsky, M.A.; Allis, C.D. Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc. Natl. Acad. Sci. USA 1998, 95, 7480–7484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prigent, C.; Dimitrov, S. Phosphorylation of serine 10 in histone H3, what for? J. Cell. Sci. 2003, 116, 3677–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendzel, M.J.; Wei, Y.; Mancini, M.A.; Van Hooser, A.; Ranalli, T.; Brinkley, B.R.; Bazett-Jones, D.P.; Allis, C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997, 106, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell. Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Blink, E.; Maianski, N.A.; Alnemri, E.S.; Zervos, A.S.; Roos, D.; Kuijpers, T.W. Intramitochondrial serine protease activity of Omi/HtrA2 is required for caspase-independent cell death of human neutrophils. Cell Death Differ. 2004, 11, 937–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilenti, L.; Lee, Y.; Hess, S.; Srinivasula, S.; Park, K.M.; Junqueira, D.; Davis, H.; Bonventre, J.V.; Alnemri, E.S.; Zervos, A.S. Characterization of a novel and specific inhibitor for the pro-apoptotic protease Omi/HtrA2. J. Biol. Chem. 2003, 278, 11489–11494. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Song, M.G.; Lee, J.S.; Kim, J.W.; Jin, J.O.; Park, J.I.; Chang, Y.C.; Kwak, J.Y. Apoptosis of human neutrophils induced by protein phosphatase 1/2A inhibition is caspase-independent and serine protease-dependent. J. Cell. Physiol. 2007, 212, 450–462. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, P.; Gómez-Martínez, A.; Carrato, A.; Martínez-Lacaci, I.; Barberá, V.M.; Soto, J.L.; Carrasco-García, E.; Menéndez-Gutierrez, M.P.; Castro-Galache, M.D.; Ferragut, J.A.; et al. Histone deacetylase inhibitors induced caspase-independent apoptosis in human pancreatic adenocarcinoma cell lines. Mol. Cancer Ther. 2005, 4, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Tiffany-Castiglioni, E.; Koh, H.C.; Son, I.H. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxicol. Lett. 2009, 191, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; Jeong, Y.; et al. Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nistér, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar] [PubMed]
- Kiess, W.; Lee, L.; Graham, D.E.; Greenstein, L.; Tseng, L.Y.; Rechler, M.M.; Nissley, S.P. Rat C6 glial cells synthesize insulin-like growth factor I (IGF-I) and express IGF-I receptors and IGF-II/mannose 6-phosphate receptors. Endocrinology 1989, 124, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Trojan, J.; Blossey, B.K.; Johnson, T.R.; Rudin, S.D.; Tykocinski, M.; Ilan, J. Loss of tumorigenicity of rat glioblastoma directed by episome-based antisense cDNA transcription of insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1992, 89, 4874–4878. [Google Scholar] [CrossRef] [PubMed]
- Strömberg, T.; Ekman, S.; Girnita, L.; Dimberg, L.Y.; Larsson, O.; Axelson, M.; Lennartsson, J.; Hellman, U.; Carlson, K.; Österborg, A.; et al. IGF-1 receptor tyrosine kinase inhibition by the cyclolignan PPP induces G2/M-phase accumulation and apoptosis in multiple myeloma cells. Blood 2006, 107, 669–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Choy, E.; Harmon, D.; Yang, C.; Ryu, K.; Schwab, J.; Mankin, H.; Hornicek, F.J. Insulin-like growth factor-I receptor tyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines. Mol. Cancer Ther. 2009, 8, 2122–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karasic, T.B.; Hei, T.K.; Ivanov, V.N. Disruption of IGF-1R signaling increases TRAIL-induced apoptosis: A new potential therapy for the treatment of melanoma. Exp. Cell Res. 2010, 316, 1994–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima-Hosoyama, S.; Hosoyama, T.; Nelon, L.D.; Keller, C. IGF-1 receptor inhibition by picropodophyllin in medulloblastoma. Biochem. Biophys. Res. Commun. 2010, 399, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Shinozaki, F.; Sugiyama, T.; Yamamoto, S.; Sueishi, M.; Yoshida, T. Insulin-like growth factor-I receptor in proliferation and motility of pancreatic cancer. World J. Gastroenterol. 2010, 16, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Doghman, M.; Axelson, M.; Lalli, E. Potent inhibitory effect of the cyclolignan picropodophyllin (PPP) on human adrenocortical carcinoma cells proliferation. Am. J. Cancer Res. 2011, 1, 356–361. [Google Scholar] [PubMed]
- E, C.; Li, J.; Shao, D.; Zhang, D.; Pan, Y.; Chen, L.; Zhang, X. The insulin-like growth factor-I receptor inhibitor picropodophyllin-induced selective apoptosis of hepatocellular carcinoma cell through a caspase-dependent mitochondrial pathway. Oncol. Res. 2013, 21, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yao, M.; Wang, L.; Yan, X.; Gu, X.; Shi, Y.; Yao, N.; Qiu, L.; Wu, W.; Yao, D. Abnormal expression of insulin-like growth factor-I receptor in hepatoma tissue and its inhibition to promote apoptosis of tumor cells. Tumour. Biol. 2013, 34, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell. Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Niikura, Y.; Dixit, A.; Scott, R.; Perkins, G.; Kitagawa, K. BUB1 mediation of caspase-independent mitotic death determines cell fate. J. Cell Biol. 2007, 178, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosengren, L.; Vasilcanu, D.; Vasilcanu, R.; Fickenscher, S.; Sehat, B.; Natalishvili, N.; Naughton, S.; Yin, S.; Girnita, A.; Girnita, L.; et al. IGF-1R tyrosine kinase expression and dependency in clones of IGF-1R knockout cells (R–). Biochem. Biophys. Res. Commun. 2006, 347, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Diepstra, A.; Xu, C.; van Imhoff, G.; Plattel, W.; Van Den Berg, A.; Visser, L. Insulin-like growth factor 1 receptor is a prognostic factor in classical Hodgkin lymphoma. PLoS ONE 2014, 9, e87474. [Google Scholar] [CrossRef] [PubMed]
- Waraky, A.; Akopyan, K.; Parrow, V.; Strömberg, T.; Axelson, M.; Abrahmsén, L.; Lindqvist, A.; Larsson, O.; Aleem, E. Picropodophyllin causes mitotic arrest and catastrophe by depolymerizing microtubules via Insulin-like growth factor-1 receptor-independent mechanism. Oncotarget 2014, 5, 8379–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta 2009, 1793, 1516–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Yamada, H.; Moriya, S.; Miyazawa, K. The beta-carboline alkaloid harmol induces cell death via autophagy but not apoptosis in human non-small cell lung cancer A549 cells. Biol. Pharm. Bull. 2011, 34, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, E. Does cancer therapy trigger cell suicide? Science 1999, 286, 2256–2258. [Google Scholar] [CrossRef] [PubMed]
- Carapancea, M.; Cosaceanu, D.; Budiu, R.; Kwiecinska, A.; Tataranu, L.; Ciubotaru, V.; Alexandru, O.; Banita, M.; Pisoschi, C.; Bäcklund, M.L.; et al. Dual targeting of IGF-1R and PDGFR inhibits proliferation in high-grade gliomas cells and induces radiosensitivity in JNK-1 expressing cells. J. Neurooncol. 2007, 85, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Bielen, A.; Perryman, L.; Box, G.M.; Valenti, M.; De Haven Brandon, A.; Martins, V.; Jury, A.; Popov, S.; Gowan, S.; Jeay, S.; et al. Enhanced efficacy of IGF1R inhibition in paediatric glioblastoma by combinatorial targeting of PDGFRα/β. Mol. Cancer Ther. 2011, 10, 1407–1418. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco-Garcia, E.; Martinez-Lacaci, I.; Mayor-López, L.; Tristante, E.; Carballo-Santana, M.; García-Morales, P.; Ventero Martin, M.P.; Fuentes-Baile, M.; Rodriguez-Lescure, Á.; Saceda, M. PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines. Cells 2018, 7, 131. https://doi.org/10.3390/cells7090131
Carrasco-Garcia E, Martinez-Lacaci I, Mayor-López L, Tristante E, Carballo-Santana M, García-Morales P, Ventero Martin MP, Fuentes-Baile M, Rodriguez-Lescure Á, Saceda M. PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines. Cells. 2018; 7(9):131. https://doi.org/10.3390/cells7090131
Chicago/Turabian StyleCarrasco-Garcia, Estefania, Isabel Martinez-Lacaci, Leticia Mayor-López, Elena Tristante, Mar Carballo-Santana, Pilar García-Morales, Maria Paz Ventero Martin, Maria Fuentes-Baile, Álvaro Rodriguez-Lescure, and Miguel Saceda. 2018. "PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines" Cells 7, no. 9: 131. https://doi.org/10.3390/cells7090131
APA StyleCarrasco-Garcia, E., Martinez-Lacaci, I., Mayor-López, L., Tristante, E., Carballo-Santana, M., García-Morales, P., Ventero Martin, M. P., Fuentes-Baile, M., Rodriguez-Lescure, Á., & Saceda, M. (2018). PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines. Cells, 7(9), 131. https://doi.org/10.3390/cells7090131