NFKB1 and Cancer: Friend or Foe?


Abstract
:1. Introduction
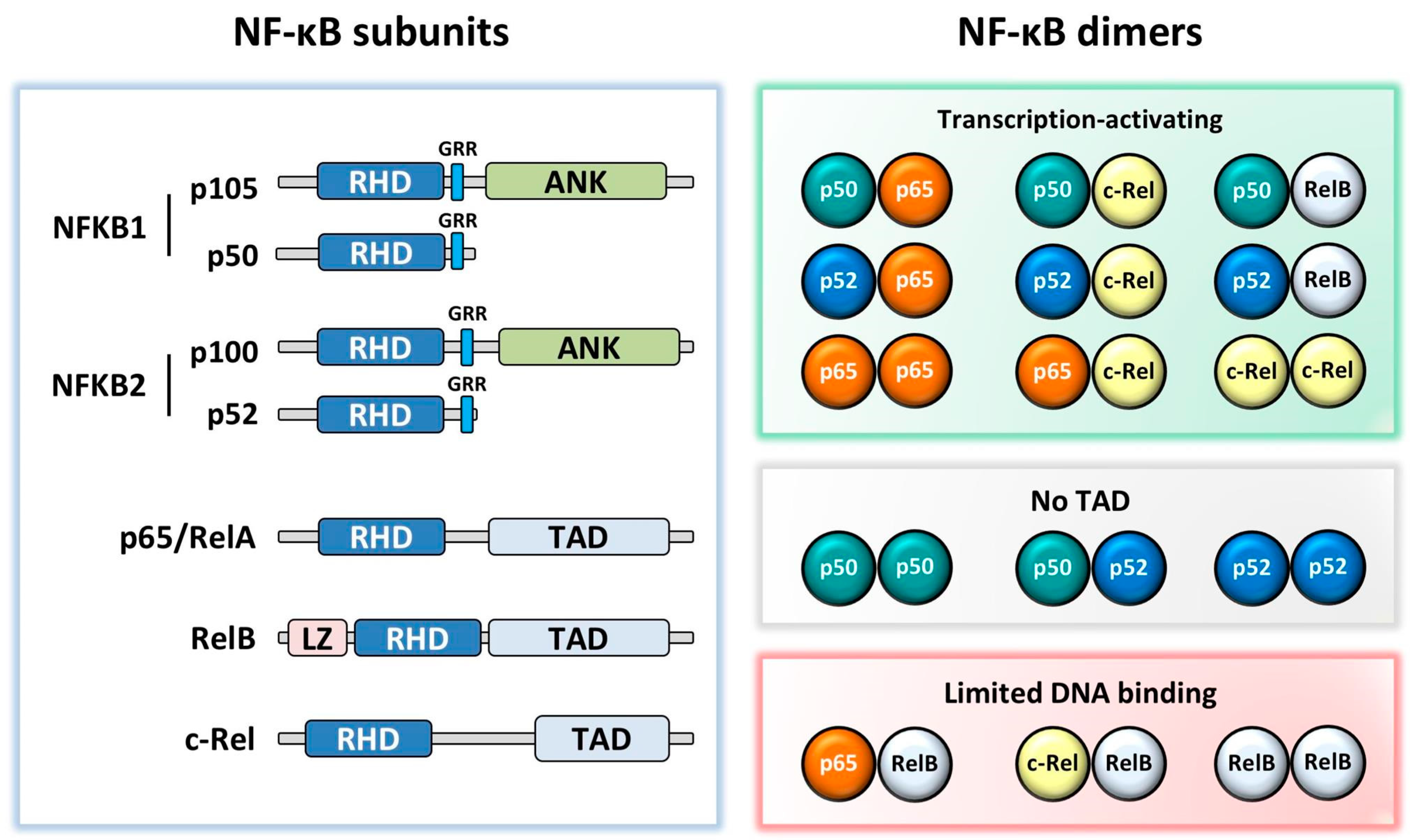
2. NF-κB Subunits
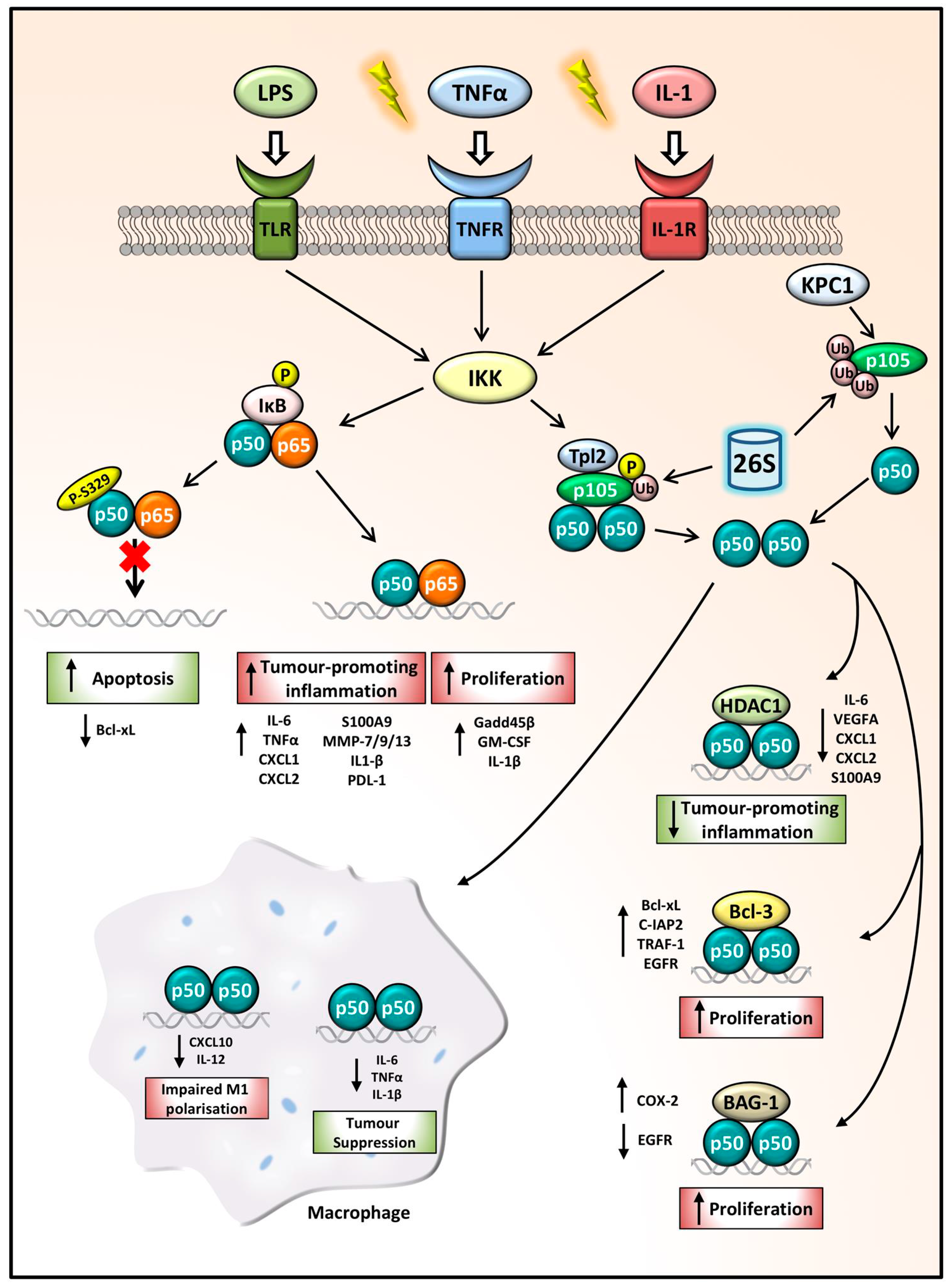
3. NFKB1 Processing
4. Co-Factor Recruitment
5. NFKB1 Tumour Suppressor
5.1. Liver Cancer
5.2. Gastric Cancer
5.3. Lung Cancer
5.4. Haematological Malignancies
6. NFKB1 Tumour Promoter
6.1. Bcl-3-Associated Cancers
6.2. Breast and Gynaecological Cancers
6.3. Colorectal and Pancreatic Cancers
6.4. Haematological Malignancies
7. NFKB1 Polymorphisms in Cancer
8. Potential Interventions
9. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALCL | anaplastic large-cell lymphoma |
| BAG-1 | BCL-2 associated athanogene |
| Bcl-3 | B-cell lymphoma 3 |
| C/EBP | CCAAT enhancer-binding protein |
| EBV | Epstein–Barr virus |
| EGFR | epidermal growth factor receptor |
| EMSA | electrophoresis mobility shift assay |
| ERK | extracellular regulated kinase |
| HCC | hepatocellular carcinoma |
| HDAC1 | histone deacetylase 1 |
| HPV16 | human papilloma virus 16 |
| IFN | interferon |
| IKK | IκB kinase |
| IL | interleukin |
| IκB | inhibitor of kappa light polypeptide gene enhancer in B-cells |
| JAK-STAT | Janus kinase-Signal transducer and activator of transcription |
| KPC1 | Kip1 ubiquitination-promoting complex 1 |
| LPS | lipopolysaccharide |
| MEK | mitogen-activated protein kinase kinase |
| NFKB1 | nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 |
| NF-κB | nuclear factor of kappa light polypeptide gene enhancer in B-cells |
| NSCLC | non-small-cell lung carcinoma |
| PD-L1 | programmed death-ligand 1 |
| PTEN | phosphatase and tensin homolog |
| SCCHN | squamous cell carcinoma of the head and neck |
| STAT | signal transducer and activator of transcription |
| TAD | transcriptional activation domain |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TNFα | tumour necrosis factor α |
| TPL-2 | tumour progression locus 2 |
References
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yokosuka, O.; Nagao, K.; Saisho, H. State of hepatitis C viral replication enhances activation of NF-kB- and AP-1-signaling induced by hepatitis B virus X. Cancer Lett. 2006, 234, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chung, G.T.-Y.; Lou, W.P.-K.; Chow, C.; To, K.-F.; Choy, K.-W.; Leung, A.W.-C.; Tong, C.Y.-K.; Yuen, J.W.-F.; Ko, C.-W.; Yip, T.T.-C.; et al. Constitutive activation of distinct NF-κB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa B kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Tchenio, T.; Piton, G.; Romeo, P.H.; Baud, V. RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 14635–14640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marienfeld, R.; May, M.J.; Berberich, I.; Serfling, E.; Ghosh, S.; Neumann, M. RelB forms transcriptionally inactive complexes with RelA/p65. J. Biol. Chem. 2003, 278, 19852–19860. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Murty, V.V.; Dalla-Favera, R.; Chaganti, R.S. Chromosomal localization of genes encoding the transcription factors, c-rel, NF-kappa Bp50, NF-kappa Bp65, and lyt-10 by fluorescence in situ hybridization. Oncogene 1993, 8, 191–193. [Google Scholar] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Sriskantharajah, S.; Belich, M.P.; Papoutsopoulou, S.; Janzen, J.; Tybulewicz, V.; Seddon, B.; Ley, S.C. Proteolysis of NF-kappaB1 p105 is essential for T cell antigen receptor-induced proliferation. Nat. Immunol. 2009, 10, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Schweighoffer, E.; Visekruna, A.; Papoutsopoulou, S.; Janzen, J.; Zillwood, R.; Tarlinton, D.M.; Tybulewicz, V.L.J.; Ley, S.C. IKK-induced NF-κB1 p105 proteolysis is critical for B cell antibody responses to T cell–dependent antigen. J. Exp. Med. 2014, 211, 2085–2101. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Robinson, M.J.; Hugunin, M.; Ley, S.C. Lipopolysaccharide Activation of the TPL-2/MEK/Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase Cascade Is Regulated by IκB Kinase-Induced Proteolysis of NF-κB1 p105. Mol. Cell. Biol. 2004, 24, 9658–9667. [Google Scholar] [CrossRef] [PubMed]
- Gantke, T.; Sriskantharajah, S.; Sadowski, M.; Ley, S.C. IκB kinase regulation of the TPL-2/ERK MAPK pathway. Immunol. Rev. 2012, 246, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Oakley, F.; Lin, F.; Packham, G.; Mann, D.A.; Mann, J. The NF-κB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 2010, 53, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-P.; Vancurova, I. Bcl3 regulates pro-survival and pro-inflammatory gene expression in cutaneous T-cell lymphoma. Biochim. Biophys. Acta 2014, 1843, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Kiely, P.A.; Carmody, R.J. Inhibition of transcription by B cell Leukemia 3 (Bcl-3) protein requires interaction with nuclear factor kappaB (NF-kappaB) p50. J. Biol. Chem. 2014, 289, 7059–7067. [Google Scholar] [CrossRef] [PubMed]
- Wessells, J.; Baer, M.; Young, H.A.; Claudio, E.; Brown, K.; Siebenlist, U.; Johnson, P.F. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J. Biol. Chem. 2004, 279, 49995–50003. [Google Scholar] [CrossRef] [PubMed]
- Paz-Priel, I.; Houng, S.; Dooher, J.; Friedman, A.D. C/EBPα and C/EBPα oncoproteins regulate nfkb1 and displace histone deacetylases from NF-κB p50 homodimers to induce NF-κB target genes. Blood 2011, 117, 4085–4094. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Buerki, C.; Lombardi, C.; Imhof, R.; Hottiger, M.O. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J. Biol. Chem. 2003, 278, 45145–45153. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Jurk, D.; Fullard, N.; Banks, P.; Page, A.; Luli, S.; Elsharkawy, A.M.; Gieling, R.G.; Chakraborty, J.B.; Fox, C.; et al. NFkappaB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015, 6, 6818. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Crawley, C.D.; Kang, S.; Raleigh, D.R.; Yu, X.; Wahlstrom, J.S.; Voce, D.J.; Darga, T.E.; Weichselbaum, R.R.; Yamini, B. p50 (NF-κB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol. Cell 2011, 44, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I.; et al. KPC1-mediated ubiquitination and proteasomal processing of NF-kappaB1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.S.; Marino, M.W.; Takahashi, T.; Yoshida, T.; Sakakura, T.; Old, L.J.; Obata, Y. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc. Natl. Acad. Sci. USA 1999, 96, 2994–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, S.; Kamata, H.; Luo, J.-L.; Leffert, H.; Karin, M. IKKβ Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation that Promotes Chemical Hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margetts, J.; Ogle, L.F.; Chan, S.L.; Chan, A.W.H.; Chan, K.C.A.; Jamieson, D.; Willoughby, C.E.; Mann, D.A.; Wilson, C.L.; Manas, D.M.; et al. Neutrophils: Driving progression and poor prognosis in hepatocellular carcinoma? Br. J. Cancer 2017, 118, 248. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Chang, P.Y.; Chen, B.S. Investigating the mechanism of hepatocellular carcinoma progression by constructing genetic and epigenetic networks using NGS data identification and big database mining method. Oncotarget 2016, 7, 79453–79473. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, H.; Yasuda, J.; Nakanishi, K.; Chuma, M.; Kamiyama, T.; Todo, S.; Hirohashi, S.; Sakamoto, M. Clinicopathological significance of nuclear factor-kappaB activation in hepatocellular carcinoma. Hepatol. Res. 2011, 41, 240–249. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Farrah, H.; Kelly, H.; Baldwin, A.S.; Funkhouser, W.K. Analysis of NF-kappa B in hepatocellular carcinoma (HCC) reveals frequent activation of p50 and bcl-3. J. Clin. Oncol. 2005, 23 (Suppl. 16), 9621. [Google Scholar] [CrossRef]
- Sokolova, O.; Naumann, M. NF-κB Signaling in Gastric Cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Takaishi, S.; Muthupalani, S.; Pritchard, D.M.; Whary, M.T.; Rogers, A.B.; Fox, J.G.; Betz, K.S.; Kaestner, K.H.; Karin, M.; et al. Conditional deletion of IkappaB-kinase beta (IKKβ) accelerates Helicobacter-dependent gastric apoptosis, proliferation and preneoplasia. Gastroenterology 2010, 138, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.A.; Putoczki, T.L.; Mielke, L.A.; Low, J.T.; Lin, A.; Preaudet, A.; Herold, M.J.; Yaprianto, K.; Tai, L.; Kueh, A.; et al. Loss of NF-κB1 Causes Gastric Cancer with Aberrant Inflammation and Expression of Immune Checkpoint Regulators in a STAT-1-Dependent Manner. Immunity 2018, 48, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, T.; Tahara, T.; Shiroeda, H.; Yamada, K.; Nomura, T.; Yamada, H.; Hayashi, R.; Matsunaga, K.; Otsuka, T.; Nakamura, M.; et al. Functional promoter polymorphisms of NFKB1 influence susceptibility to the diffuse type of gastric cancer. Oncol. Rep. 2013, 30, 3013–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, T.; Qinsheng, W.; Xuxia, W.; Shuguang, Z.; Ming, Q.; Zhenxiong, L.; Jingjie, W. Nuclear Factor-Kappa B1 is Associated With Gastric Cancer in a Chinese Population. Medicine 2014, 93, e279. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Kang, W.; Zhang, B.; Wu, F.; Dong, Y.; Tong, J.H.M.; Yang, W.; Zhou, Y.; Zhang, L.; Cheng, A.S.L.; et al. miR-508-3p concordantly silences NFKB1 and RELA to inactivate canonical NF-κB signaling in gastric carcinogenesis. Mol. Cancer 2016, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Qu, Z.; Xiao, Y.; Zhou, J.; Burns, T.F.; Stabile, L.P.; Siegfried, J.M.; Xiao, G. NF-kappaB1 p105 suppresses lung tumorigenesis through the Tpl2 kinase but independently of its NF-kappaB function. Oncogene 2016, 35, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Al-Saad, S.; Al-Shibli, K.; Donnem, T.; Persson, M.; Bremnes, R.M.; Busund, L.T. The prognostic impact of NF-κB p105, vimentin, E-cadherin and Par6 expression in epithelial and stromal compartment in non-small-cell lung cancer. Br. J. Cancer 2008, 99, 1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.; Li, C.; Huang, C.; Zhuang, W.; Huang, Y.; Xu, H.; Miao, Q.; Hu, D. Co-expression of NF-kappaB-p65 and phosphorylated NF-kappaB-p105 is associated with poor prognosis in surgically resectable non-small cell lung cancer. J. Cell. Mol. Med. 2018, 22, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Voce, D.J.; Schmitt, A.M.; Uppal, A.; McNerney, M.E.; Bernal, G.M.; Cahill, K.E.; Wahlstrom, J.S.; Nassiri, A.; Yu, X.; Crawley, C.D.; et al. Nfkb1 is a haploinsufficient DNA damage-specific tumor suppressor. Oncogene 2015, 34, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Crawley, C.D.; Kang, S.; Bernal, G.M.; Wahlstrom, J.S.; Voce, D.J.; Cahill, K.E.; Garofalo, A.; Raleigh, D.R.; Weichselbaum, R.R.; Yamini, B. S-phase-dependent p50/NF-кB1 phosphorylation in response to ATR and replication stress acts to maintain genomic stability. Cell Cycle 2015, 14, 566–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Fujii, M.; Ikeda, S.; Yamada, Y.; Tomonaga, M.; Ballard, D.W.; Yamamoto, N. Constitutive Activation of NF-κB in Primary Adult T-Cell Leukemia Cells. Blood 1999, 93, 2360–2368. [Google Scholar] [PubMed]
- Mathas, S.; Johrens, K.; Joos, S.; Lietz, A.; Hummel, F.; Janz, M.; Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Lichter, P.; et al. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005, 106, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Budunova, I.V.; Perez, P.; Vaden, V.R.; Spiegelman, V.S.; Slaga, T.J.; Jorcano, J.L. Increased expression of p50-NF-kappaB and constitutive activation of NF-kappaB transcription factors during mouse skin carcinogenesis. Oncogene 1999, 18, 7423–7431. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bharti, A.C.; Varghese, P.; Saluja, D.; Das, B.C. Differential expression and activation of NF-kappaB family proteins during oral carcinogenesis: Role of high risk human papillomavirus infection. Int. J. Cancer 2006, 119, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, N.J.; Pathmanathan, R.; Raab-Traub, N. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res. 2003, 63, 8293–8301. [Google Scholar] [PubMed]
- Cogswell, P.C.; Guttridge, D.C.; Funkhouser, W.K.; Baldwin, A.S., Jr. Selective activation of NF-kappa B subunits in human breast cancer: Potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene 2000, 19, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Eppenberger-Castori, S.; Marx, C.; Yau, C.; Scott, G.K.; Eppenberger, U.; Benz, C.C. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int. J. Biochem. Cell. Biol. 2005, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Rosenthal, D.; Perec, L.; Merajver, S. Down regulation of NfkB1 expression blocks cell motility in a cell line model of inflammatory breast cancer. Cancer Res. 2008, 68, 1991. [Google Scholar]
- Havard, L.; Rahmouni, S.; Boniver, J.; Delvenne, P. High levels of p105 (NFKB1) and p100 (NFKB2) proteins in HPV16-transformed keratinocytes: Role of E6 and E7 oncoproteins. Virology 2005, 331, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Xie, L.; Wang, X.; He, H.; Lin, Y.; Hu, L. Association of constitutive nuclear factor-kappaB activation with aggressive aspects and poor prognosis in cervical cancer. Int. J. Gynecol. Cancer 2009, 19, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Prusty, B.K.; Husain, S.A.; Das, B.C. Constitutive activation of nuclear factor -kB: Preferntial homodimerization of p50 subunits in cervical carcinoma. Front. Biosci. 2005, 10, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Giopanou, I.; Bravou, V.; Papanastasopoulos, P.; Lilis, I.; Aroukatos, P.; Papachristou, D.; Kounelis, S.; Papadaki, H. Metadherin, p50, and p65 expression in epithelial ovarian neoplasms: An immunohistochemical study. Biomed. Res. Int. 2014, 2014, 178410. [Google Scholar] [CrossRef] [PubMed]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Ippolito, A.; Consonni, F.M.; Carraro, L.; Celesti, G.; Correale, C.; Grizzi, F.; Pasqualini, F.; Tartari, S.; Rinaldi, M.; et al. Protumor Steering of Cancer Inflammation by p50 NF-κB Enhances Colorectal Cancer Progression. Cancer Immunol. Res. 2018, 6, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Southern, S.L.; Collard, T.J.; Urban, B.C.; Skeen, V.R.; Smartt, H.J.; Hague, A.; Oakley, F.; Townsend, P.A.; Perkins, N.D.; Paraskeva, C.; et al. BAG-1 interacts with the p50-p50 homodimeric NF-kappaB complex: Implications for colorectal carcinogenesis. Oncogene 2012, 31, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, J.S.; Kim, W.K.; Oh, K.J.; Kim, J.M.; Lee, H.J.; Han, B.S.; Kim, D.S.; Seo, Y.S.; Lee, S.C.; et al. Intracellular annexin A2 regulates NF-kappaB signaling by binding to the p50 subunit: Implications for gemcitabine resistance in pancreatic cancer. Cell Death Dis. 2015, 6, e1606. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.; Garg, B.; Modi, S.; George, J.; Sethi, V.; Banerjee, S.; Saluja, A.; Dudeja, V. NF-κB P50 Subunit in Stellate-Cells Actively Modulates Cancer Growth in Mouse Models of Pancreatic Cancer. J. Am. Coll. Surg. 2016, 223, S142. [Google Scholar] [CrossRef]
- Chen, T.L.; Tran, M.; Lakshmanan, A.; Harrington, B.K.; Goettl, V.M.; Lehman, A.M.; Trudeau, S.; Lucas, D.M.; Johnson, A.J.; Byrd, J.C.; et al. NF-κB p50 (nfkb1) contributes to pathogenesis in the Eμ-TCL1 mouse model of chronic lymphocytic leukemia. Blood 2017, 130, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.C.; Liou, H.-C.; Tuomanen, E.I.; Baltimore, D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell 1995, 80, 321–330. [Google Scholar] [CrossRef]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Koff, J.L.; Moffitt, A.B.; Cinar, M.; Ramachandiran, S.; Chen, Z.; Switchenko, J.M.; Mosunjac, M.; Neill, S.G.; Mann, K.P.; et al. Molecular impact of selective NFKB1 and NFKB2 signaling on DLBCL phenotype. Oncogene 2017, 36, 4224–4232. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Tu, M.; Xu-Monette, Z.Y.; Sun, R.; Manyam, G.C.; Xu, X.; Tzankov, A.; His, E.D.; Møller, M.B.; Medeiros, L.J.; et al. NF-κB p50 activation associated with immune dysregulation confers poorer survival for diffuse large B-cell lymphoma patients with wild-type p53. Mod. Pathol. 2017, 30, 854. [Google Scholar] [CrossRef] [PubMed]
- Yenmis, G.; Oner, T.; Cam, C.; Koc, A.; Kucuk, O.S.; Yakicier, M.C.; Dizman, D.; Sultuybek, G.K. Association of NFKB1 and NFKBIA polymorphisms in relation to susceptibility of Behcet’s disease. Scand. J. Immunol. 2015, 81, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Koc, A.; Batar, B.; Celik, O.; Onaran, I.; Tasan, E.; Sultuybek, G.K. Polymorphism of the NFKB1 affects the serum inflammatory levels of IL-6 in Hashimoto thyroiditis in a Turkish population. Immunobiology 2014, 219, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Lewander, A.; Butchi, A.K.; Gao, J.; He, L.J.; Lindblom, A.; Arbman, G.; Carstensen, J.; Zhang, Z.Y.; Sun, X.F. Polymorphism in the promoter region of the NFKB1 gene increases the risk of sporadic colorectal cancer in Swedish but not in Chinese populations. Scand. J. Gastroenterol. 2007, 42, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Mohd Suzairi, M.S.; Tan, S.C.; Ahmad Aizat, A.A.; Mohd Aminudin, M.; Siti Nurfatimah, M.S.; Andee, Z.D.; Ankathil, R. The functional -94 insertion/deletion ATTG polymorphism in the promoter region of NFKB1 gene increases the risk of sporadic colorectal cancer. Cancer Epidemiol. 2013, 37, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Su, J.-L.; Lin, C.-W.; Su, C.-W.; Shih, C.-H.; Yang, S.-F.; Chien, M.-H. Effects of NFKB1 and NFKBIA Gene Polymorphisms on Hepatocellular Carcinoma Susceptibility and Clinicopathological Features. PLoS ONE 2013, 8, e56130. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gu, J.; Yang, X.; Cai, H.; Tao, J.; Yang, X.; Lu, Q.; Wang, Z.; Yin, C.; Gu, M. Functional promoter -94 ins/del ATTG polymorphism in NFKB1 gene is associated with bladder cancer risk in a Chinese population. PLoS ONE 2013, 8, e71604. [Google Scholar] [CrossRef] [PubMed]
- Umar, M.; Upadhyay, R.; Kumar, S.; Ghoshal, U.C.; Mittal, B. Association of Common Polymorphisms in TNFA, NFkB1 and NFKBIA with Risk and Prognosis of Esophageal Squamous Cell Carcinoma. PLoS ONE 2013, 8, e81999. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, H.; Liang, Y.; Sun, R.; Wei, T.; Li, Z.; Gong, Y.; Gong, R.; Liu, F.; Zhang, L.; et al. A Functional Insertion/Deletion Polymorphism in the Promoter Region of the NFKB1 Gene Increases the Risk of Papillary Thyroid Carcinoma. Genet. Test. Mol. Biomark. 2015, 19, 167–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, G.F.; Arraes, J.A.; Bakos, L.; Ashton-Prolla, P.; Giugliani, R.; Callegari-Jacques, S.M.; Santos, S.; Bakos, R.M. Polymorphisms in CYP19A1 and NFKB1 genes are associated with cutaneous melanoma risk in southern Brazilian patients. Melanoma Res. 2016, 26, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.; Rosdahl, I.; Sun, X.F.; Zhang, H. Importance of polymorphisms in NF-kappaB1 and NF-kappaBIalpha genes for melanoma risk, clinicopathological features and tumor progression in Swedish melanoma patients. J. Cancer Res. Clin. Oncol. 2007, 133, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Rao, L.; Li, Y.; Gao, L.; Wang, Y.; Chen, Y.; Xue, H.; Song, Y.; Peng, Y.; Liao, M.; et al. A functional insertion/deletion polymorphism in the promoter region of NFKB1 gene increases susceptibility for nasopharyngeal carcinoma. Cancer Lett. 2009, 275, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Hsieh, Y.S.; Hsin, C.H.; Su, C.W.; Lin, C.H.; Wei, L.H.; Yang, S.F.; Chien, M.H. Effects of NFKB1 and NFKBIA gene polymorphisms on susceptibility to environmental factors and the clinicopathologic development of oral cancer. PLoS ONE 2012, 7, e35078. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Begin, L.R.; Gleave, M.E.; Mes-Masson, A.M.; Saad, F. Nuclear localisation of nuclear factor-kappaB transcription factors in prostate cancer: An immunohistochemical study. Br. J. Cancer 2005, 93, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Qie, M.; Wang, Y.; Yan, L.; Zhang, Z.; Liang, A.; Wang, T.; Wang, X.; Song, Y.; Zhang, L. Relationship between NFKB1 -94 insertion/deletion ATTG polymorphism and susceptibility of cervical squamous cell carcinoma risk. Ann. Oncol. 2010, 21, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Pallavi, S.; Anoop, K.; Showket, H.; Alo, N.; Mausumi, B. NFKB1/NFKBIa polymorphisms are associated with the progression of cervical carcinoma in HPV-infected postmenopausal women from rural area. Tumor Biol. 2015, 36, 6265–6276. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, H.-L.; Gao, S.; Zhang, W.; Tan, Y.-T.; Rothman, N.; Purdue, M.; Gao, Y.-T.; Zheng, W.; Shu, X.-O.; et al. Genetic polymorphism of NFKB1 and NFKBIA genes and liver cancer risk: A nested case–control study in Shanghai, China. BMJ Open 2014, 4, e004427. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, R.; Zheng, H.; Xiao, R.; Feng, J.; Wang, H.; Gao, X.; Guo, L. The NFKB1 polymorphism (rs4648068) is associated with the cell proliferation and motility in gastric cancer. BMC Gastroenterol. 2015, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Sunakawa, Y.; Stremitzer, S.; Cao, S.; Zhang, W.; Yang, D.; Wakatsuki, T.; Ning, Y.; Yamauchi, S.; Stintzing, S.; Sebio, A.; et al. Association of variants in genes encoding for macrophage-related functions with clinical outcome in patients with locoregional gastric cancer. Ann. Oncol. 2015, 26, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.P.; Cai, P.S.; Liang, H.B. Association of the genetic polymorphisms of NFKB1 with susceptibility to ovarian cancer. Genet. Mol. Res. 2015, 14, 8273–8282. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Grassia, G.; Colleran, A.; Kiely, P.A.; Ialenti, A.; Maffia, P.; Carmody, R.J. Mapping the Interaction of B Cell Leukemia 3 (BCL-3) and Nuclear Factor kappaB (NF-kappaB) p50 Identifies a BCL-3-mimetic Anti-inflammatory Peptide. J. Biol. Chem. 2015, 290, 15687–15696. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| SNP | Cancer Type | Sequence; Location; Consequence (If Known) | Prognostic | Cohort | Reference |
|---|---|---|---|---|---|
| rs28362491 | Colorectal | ATTG del; -94 promoter region; reduces promoter activity and nuclear protein binding ability, decreasing NFKB1 expression | Disadvantageous | Swedish; Malaysian | [68,69] |
| Gastric | Disadvantageous | Japanese; Chinese | [36,37] | ||
| Liver | Disadvantageous | Taiwanese | [70] | ||
| Bladder | Disadvantageous | Chinese | [71] | ||
| Esophageal | Disadvantageous | Northern Indian | [72] | ||
| Thyroid | Disadvantageous | Chinese | [73] | ||
| Melanoma | Disadvantageous | Brazilian, Swedish | [74,75] | ||
| Nasopharyngeal | Disadvantageous | Chinese | [76] | ||
| Oral | Disadvantageous | Taiwanese | [77] | ||
| Prostate | Advantageous | Canadian | [78] | ||
| Ovarian | Advantageous | Greek | [55] | ||
| Cervical | Advantageous | Chinese, Indian | [79,80] | ||
| rs230496 | Liver | AG and GG genotypes | Disadvantageous | Chinese | [81] |
| rs230525 | Liver | GA genotypes | Disadvantageous | Chinese | [81] |
| rs230530 | Liver | AA genotypes | Disadvantageous | Chinese | [81] |
| rs78696119 | Gastric | GG genotypes; increased inflammatory cell infiltration, diffuse type gastric cancer and cancer progression | Disadvantageous | Japanese | [36] |
| rs72696119 | Gastric | GG and del/del genotypes; -449 in 5′ UTR region; muscle layer tumour invasion and lymph node metastasis | Disadvantageous | Japanese | [36] |
| rs4648068 | Gastric | GG genotype; intron 12; increased incorporation of the H3K9me1 and H3K4me1 histones, increased chemomodification, enhanced transcriptional activity, cell proliferation and motility | Disadvantageous | Chinese | [37,82] |
| rs230510 | Gastric | A genotype; intronic region | Advantageous | Japanese, US | [83] |
| Ovarian | T genotype; intron 12 | Advantageous | Chinese | [84] | |
| rs230521 | Ovarian | G genotype; intron 4 | Advantageous | Chinese | [84] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Concetti, J.; Wilson, C.L. NFKB1 and Cancer: Friend or Foe? Cells 2018, 7, 133. https://doi.org/10.3390/cells7090133
Concetti J, Wilson CL. NFKB1 and Cancer: Friend or Foe? Cells. 2018; 7(9):133. https://doi.org/10.3390/cells7090133
Chicago/Turabian StyleConcetti, Julia, and Caroline L. Wilson. 2018. "NFKB1 and Cancer: Friend or Foe?" Cells 7, no. 9: 133. https://doi.org/10.3390/cells7090133
APA StyleConcetti, J., & Wilson, C. L. (2018). NFKB1 and Cancer: Friend or Foe? Cells, 7(9), 133. https://doi.org/10.3390/cells7090133