Vacuolar H+-ATPase Subunit V0C Regulates Aerobic Glycolysis of Esophageal Cancer Cells via PKM2 Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. RNA Purification and qRT-PCR
2.3. Cell Viability Assay
2.4. Western Blotting
2.5. Migration and Invasion Assays
2.6. Immunoprecipitation
2.7. GST Pull-Down Assay
2.8. Immunocytochemistry
2.9. Lactate and ATP Assay
2.10. Glucose Uptake Assay and Flow Cytometry
2.11. Annexin V Staining
2.12. Measurement of Extracellular Acidification Rate and Oxygen Consumption Rate
2.13. Subcellular Fractionation
2.14. Chromatin Immunoprecipitation Assay
2.15. Statistical Analysis
3. Results
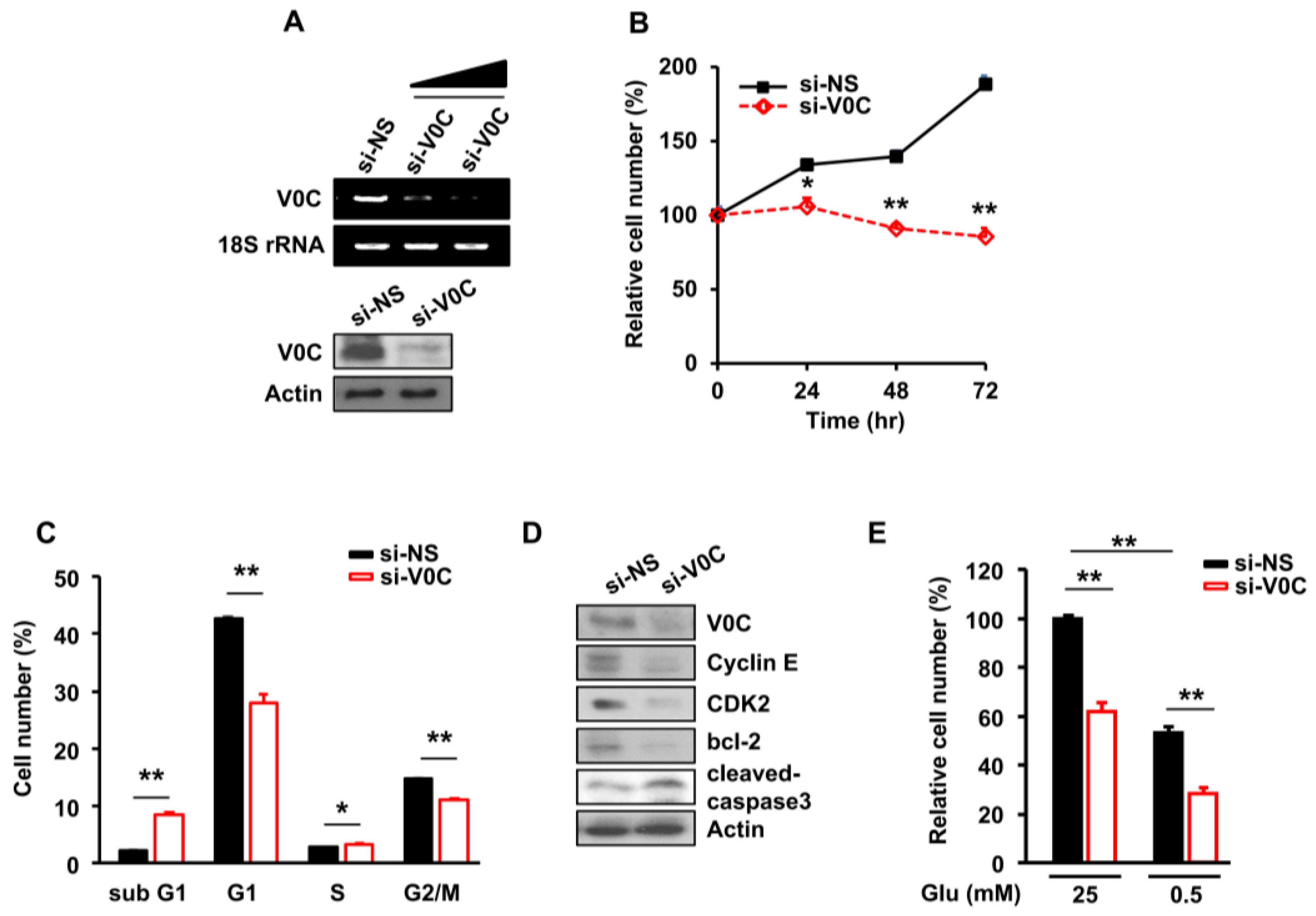
3.1. ATP6V0C Depletion Inhibits ECC Proliferation
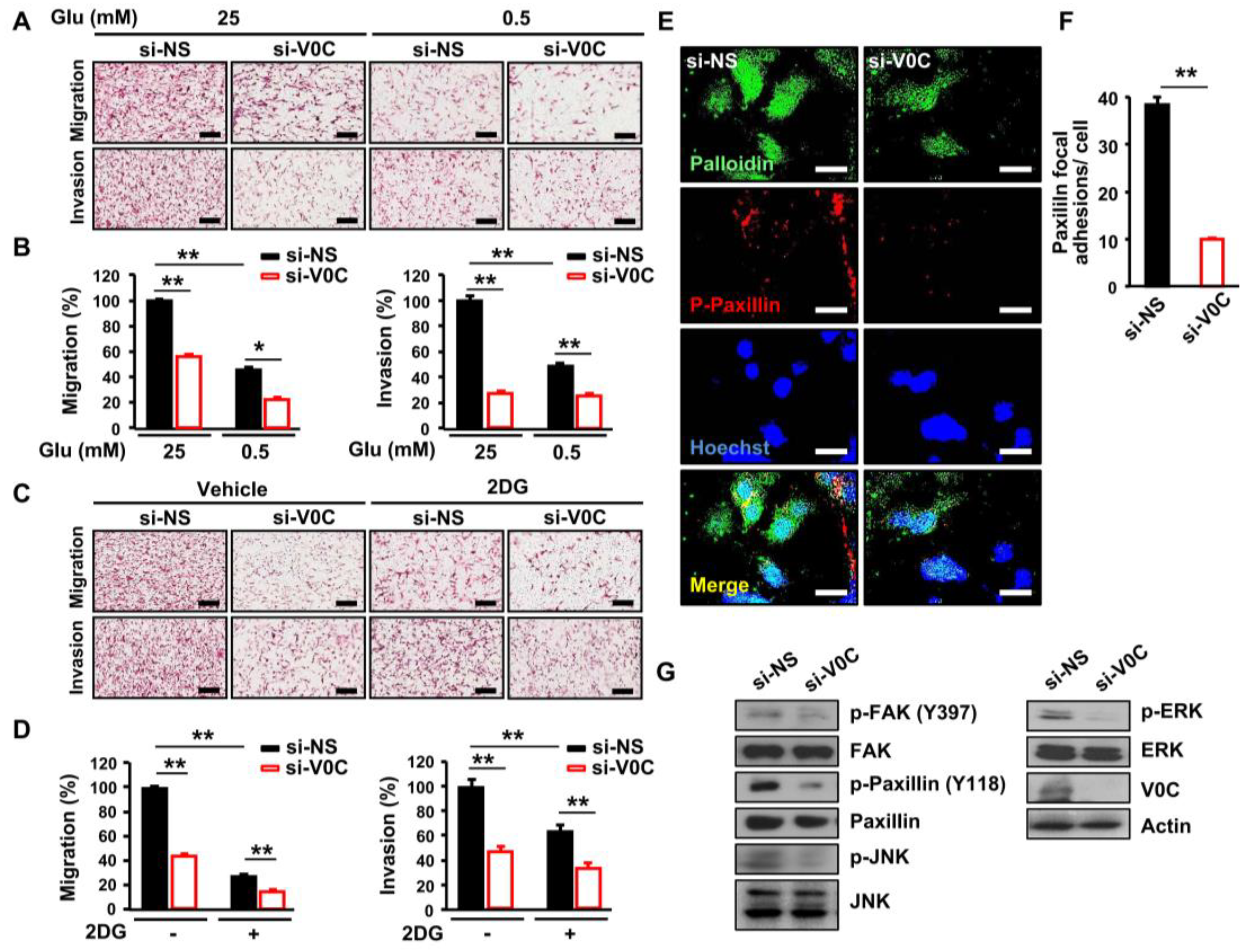
3.2. ATP6V0C Depletion Attenuates Cell Adhesion, Migration, and Invasion In ECCs
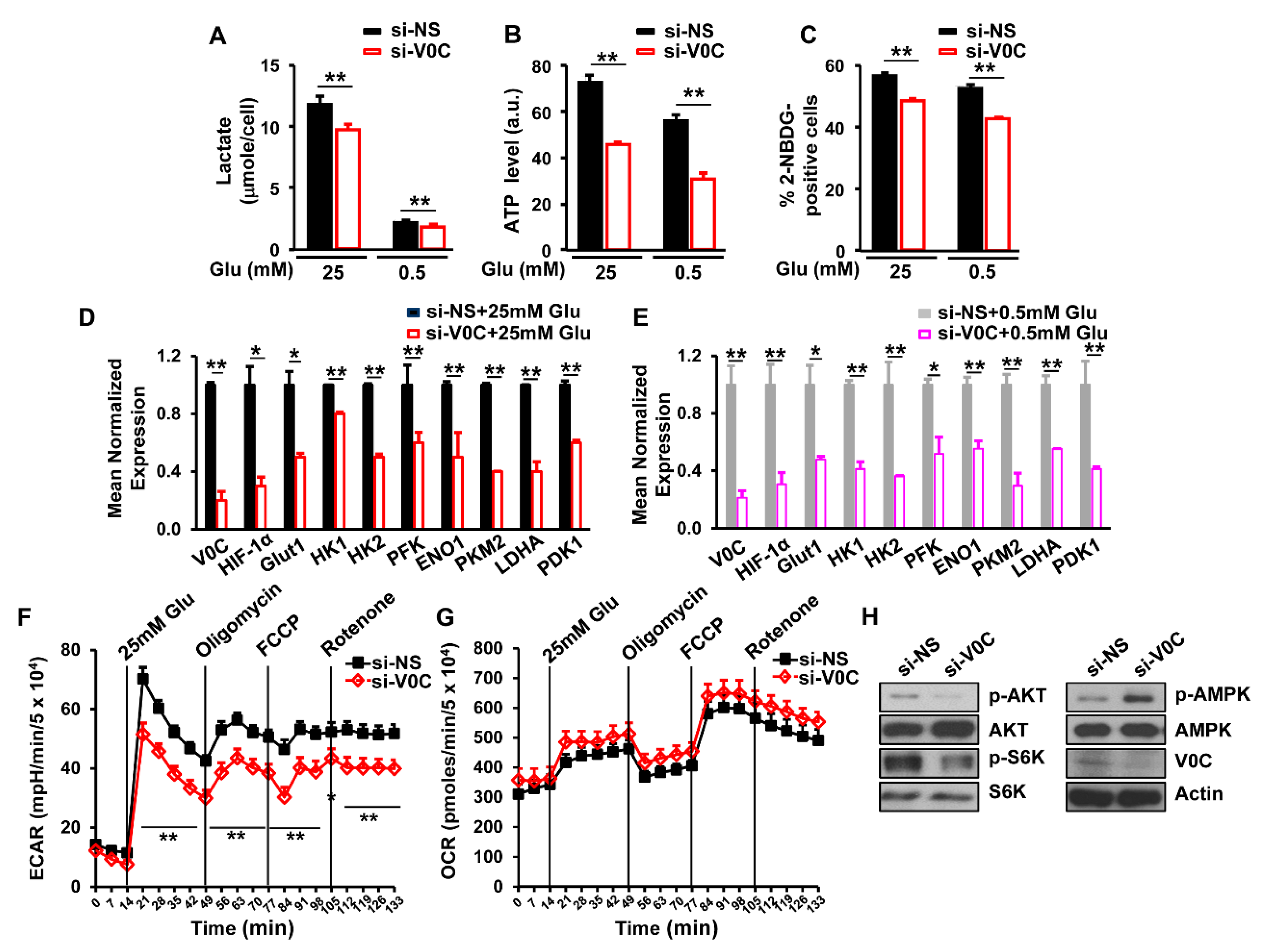
3.3. ATP6V0C Enhances Aerobic Glycolysis in ECCs
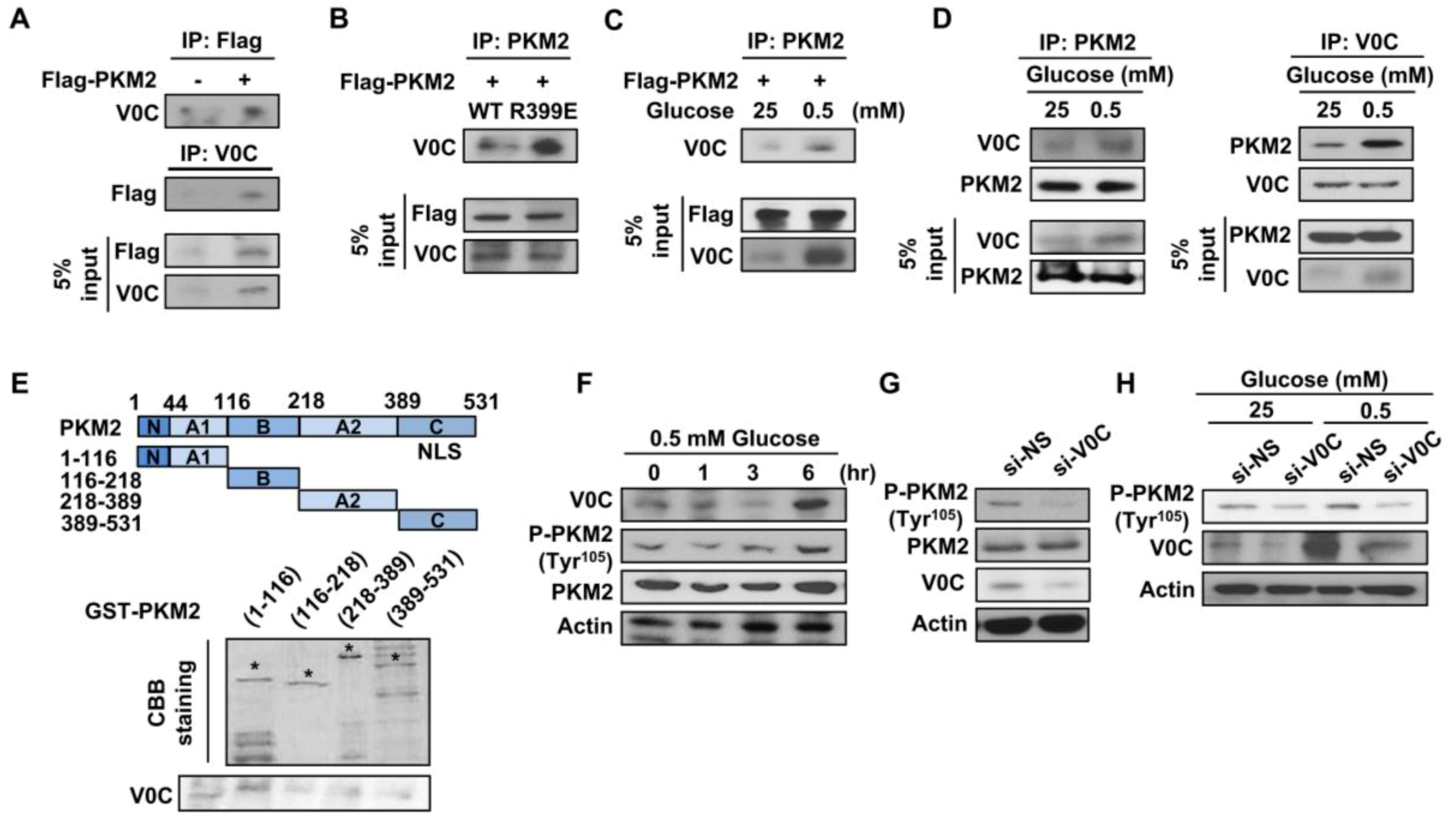
3.4. ATP6V0C Interacts with PKM2 and Increases Its Phosphorylation at Tyr105
3.5. ATP6V0C Induces Nuclear Translocation of PKM2 and Increases Glycolytic Gene Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nassri, A.; Zhu, H.; Muftah, M.; Ramzan, Z. Epidemiology and Survival of Esophageal Cancer Patients in an American Cohort. Cureus 2018, 10, 2507. [Google Scholar] [CrossRef]
- Zhang, Y. Epidemiology of esophageal cancer. World J. Gastroenterol. 2013, 19, 5598–5606. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Boil. Med. 2014, 11, 1–19. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Tohma, T.; Okazumi, S.; Makino, H.; Cho, A.; Mochiduki, R.; Shuto, K.; Kudo, H.; Matsubara, K.; Gunji, H.; Ochiai, T. Relationship between glucose transporter, hexokinase and FDG-PET in esophageal cancer. Hepatogastroenterology 2005, 52, 486–490. [Google Scholar]
- Wang, L.; Li, J.-J.; Guo, L.-Y.; Li, P.; Zhao, Z.; Zhou, H.; Di, L.-J. Molecular link between glucose and glutamine consumption in cancer cells mediated by CtBP and SIRT4. Oncogenesis 2018, 7, 26. [Google Scholar] [CrossRef]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [Green Version]
- Buller, C.L.; Heilig, C.W.; Brosius, F.C., 3rd. Glut1 enhances mtor activity independently of tsc2 and ampk. Am. J. Physiol. Renal. Physiol. 2011, 301, F588–F596. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. Mtorc1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar h(+)-atpase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [Green Version]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Boil. 2007, 8, 917–929. [Google Scholar] [CrossRef]
- Ohta, T.; Numata, M.; Yagishita, H.; Futagami, F.; Tsukioka, Y.; Kitagawa, H.; Kayahara, M.; Nagakawa, T.; Miyazaki, I.; Yamamoto, M.; et al. Expression of 16 kda proteolipid of vacuolar-type h(+)-atpase in human pancreatic cancer. Br. J. Cancer 1996, 73, 1511–1517. [Google Scholar] [CrossRef]
- Kubota, S.; Seyama, Y. Overexpression of Vacuolar ATPase 16-kDa Subunit in 10T1/2 Fibroblasts Enhances Invasion with Concomitant Induction of Matrix Metalloproteinase-2. Biochem. Biophys. Res. Commun. 2000, 278, 390–394. [Google Scholar] [CrossRef]
- Kato, H.; Takita, J.; Miyazaki, T.; Nakajima, M.; Fukai, Y.; Masuda, N.; Fukuchi, M.; Manda, R.; Ojima, H.; Tsukada, K.; et al. Glut-1 glucose transporter expression in esophageal squamous cell carcinoma is associated with tumor aggressiveness. Anticancer Res. 2002, 22, 2635–2639. [Google Scholar]
- Yeo, S.-Y.; Ha, S.-Y.; Lee, K.-W.; Cui, Y.; Yang, Z.-T.; Xuan, Y.-H.; Kim, S.-H. Twist1 is highly expressed in cancer-associated fibroblasts of esophageal squamous cell carcinoma with a prognostic significance. Oncotarget 2017, 8, 65265–65280. [Google Scholar] [CrossRef] [Green Version]
- Yeo, S.-Y.; Lee, K.-W.; Shin, D.; An, S.; Cho, K.-H.; Kim, S.-H. A positive feedback loop bi-stably activates fibroblasts. Nat. Commun. 2018, 9, 3016. [Google Scholar] [CrossRef]
- Kim, K.; Pyo, S.; Um, S.H. S6 kinase 2 deficiency enhances ketone body production and increases peroxisome proliferator-activated receptor alpha activity in the liver. Hepatology 2012, 55, 1727–1737. [Google Scholar] [CrossRef]
- Chau, G.C.; Im, D.U.; Kang, T.M.; Bae, J.M.; Kim, W.; Pyo, S.; Moon, E.Y.; Um, S.H. Mtor controls chrebp transcriptional activity and pancreatic beta cell survival under diabetic stress. J. Cell Biol. 2017, 216, 2091–2105. [Google Scholar] [CrossRef]
- Wang, L.; Han, X.; Qu, G.; Su, L.; Zhao, B.; Miao, J. A pH probe inhibits senescence in mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 343. [Google Scholar] [CrossRef]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH sensing and regulation in cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Qiu, J.J.; Zhu, S.; Cao, B.; Sun, L.; Li, S.; Li, P.; Zhang, S.; Dong, S. Oncogenic Features of PHF8 Histone Demethylase in Esophageal Squamous Cell Carcinoma. PLoS ONE 2013, 8, 77353. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouysségur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Boil. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Fei, Q.; Li, J.; Sun, Y.; Zhu, C.; Wang, F. 2-Deoxyglucose Reverses the Promoting Effect of Insulin on Colorectal Cancer Cells In Vitro. PLoS ONE 2016, 11, 0151115. [Google Scholar] [CrossRef]
- Elias, L.A.B.; Wang, D.D.; Kriegstein, A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nature 2007, 448, 901–907. [Google Scholar] [CrossRef]
- Zhao, J.; Guan, J.-L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Tong, A.; Wang, F.; Cui, Y.; Li, C.; Zhang, Y.; Yan, Z. The roles of pi3k/akt/mtor and mapk/erk signaling pathways in human pheochromocytomas. Int. J. Endocrinol. 2016, 2016, 5286972. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase—An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.-C.; Hung, W.-C.; Yamaguchi, H.; Lim, S.-O.; Liao, H.-W.; Tsai, C.-H.; Hung, M.-C. Extracellular PKM2 induces cancer proliferation by activating the EGFR signaling pathway. Am. J. Cancer Res. 2016, 6, 628–638. [Google Scholar]
- Prakasam, G.; Iqbal, M.A.; Bamezai, R.N.K.; Mazurek, S. Posttranslational Modifications of Pyruvate Kinase M2: Tweaks that Benefit Cancer. Front. Oncol. 2018, 8, 22. [Google Scholar] [CrossRef]
- Hitosugi, T.; Kang, S.; Heiden, M.G.V.; Chung, T.-W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 is a PHD3-stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar ATPase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef]
- Pavelin, J.; Reynolds, N.; Chiweshe, S.; Wu, G.; Tiribassi, R.; Grey, F. Systematic MicroRNA Analysis Identifies ATP6V0C as an Essential Host Factor for Human Cytomegalovirus Replication. PLoS Pathog. 2013, 9, e1003820. [Google Scholar] [CrossRef]
- Pavelin, J.; McCormick, D.; Chiweshe, S.; Ramachandran, S.; Lin, Y.-T.; Grey, F. Cellular v-ATPase is required for virion assembly compartment formation in human cytomegalovirus infection. Open Boil. 2017, 7, 160298. [Google Scholar] [CrossRef]
- McConnell, M.; Feng, S.; Chen, W.; Zhu, G.; Shen, D.; Ponnazhagan, S.; Deng, L.; Li, Y.-P. Osteoclast proton pump regulator Atp6v1c1 enhances breast cancer growth by activating the mTORC1 pathway and bone metastasis by increasing V-ATPase activity. Oncotarget 2017, 8, 47675–47690. [Google Scholar] [CrossRef] [Green Version]
- Avnet, S.; Di Pompo, G.; Lemma, S.; Salerno, M.; Perut, F.; Bonuccelli, G.; Granchi, D.; Zini, N.; Baldini, N. V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Kobia, F.; Duchi, S.; Deflorian, G.; Vaccari, T. Pharmacologic inhibition of vacuolar h+ atpase reduces physiologic and oncogenic notch signaling. Mol. Oncol. 2014, 8, 207–220. [Google Scholar] [CrossRef]
- Michel, V.; Licon-Munoz, Y.; Trujillo, K.; Bisoffi, M.; Parra, K.J. Inhibitors of vacuolar atpase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int. J. Cancer 2013, 132, E1–E10. [Google Scholar] [CrossRef]
- Zhang, S.; Schneider, L.S.; Vick, B.; Grunert, M.; Jeremias, I.; Menche, D.; Müller, R.; Vollmar, A.M.; Liebl, J. Anti-leukemic effects of the V-ATPase inhibitor Archazolid A. Oncotarget 2015, 6, 43508–43528. [Google Scholar] [CrossRef] [Green Version]
- Higashida, H.; Yokoyama, S.; Tsuji, C.; Muramatsu, S.I. Neurotransmitter release: Vacuolar atpase v0 sector c-subunits in possible gene or cell therapies for parkinson’s, alzheimer’s, and psychiatric diseases. J. Physiol. Sci. 2017, 67, 11–17. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Wu, C.-A.; Chao, Y.; Shiah, S.-G.; Lin, W.-W. Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1833, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Sautin, Y.Y.; Holliday, L.S.; Gluck, S.L. The glycolytic enzyme aldolase mediates assembly, expression, and activity of vacuolar h+-atpase. J. Biol. Chem. 2004, 279, 8732–8739. [Google Scholar] [CrossRef]
- Christofk, H.R.; Heiden, M.G.V.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Boil. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.-R.; Jenny, J.Y. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Li, W.; Xu, Z.; Hong, J.; Xu, Y. Expression patterns of three regulation enzymes in glycolysis in esophageal squamous cell carcinoma: Association with survival. Med. Oncol. 2014, 31, 118. [Google Scholar] [CrossRef]
- Jefferies, K.C.; Cipriano, D.J.; Forgac, M. Function, structure and regulation of the vacuolar (h+)-atpases. Arch. Biochem. Biophys. 2008, 476, 33–42. [Google Scholar] [CrossRef]
- Lu, M.; Ammar, D.; Ives, H.; Albrecht, F.; Gluck, S.L. Physical interaction between aldolase and vacuolar h+-atpase is essential for the assembly and activity of the proton pump. J. Biol. Chem. 2007, 282, 24495–24503. [Google Scholar] [CrossRef]
- Keller, K.E.; Tan, I.S.; Lee, Y.-S. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science 2012, 338, 1069–1072. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Z.; Li, M.; Yu, D. Abstract 2374: Oncogenic kinases-induced PKM2 tyrosine 105 phosphorylation converts nononcogenic PKM2 to a tumor promoter and induces cancer stem-like cells. Mol. Cell. Biol. 2018, 78, 2374. [Google Scholar] [CrossRef]
- Wang, H.J.; Hsieh, Y.J.; Cheng, W.C.; Lin, C.P.; Lin, Y.S.; Yang, S.F.; Chen, C.C.; Izumiya, Y.; Yu, J.S.; Kung, H.J.; et al. Jmjd5 regulates pkm2 nuclear translocation and reprograms hif-1alpha-mediated glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 279–284. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, S.W.; Chau, G.C.; Kim, S.-T.; Um, S.H. Vacuolar H+-ATPase Subunit V0C Regulates Aerobic Glycolysis of Esophageal Cancer Cells via PKM2 Signaling. Cells 2019, 8, 1137. https://doi.org/10.3390/cells8101137
Son SW, Chau GC, Kim S-T, Um SH. Vacuolar H+-ATPase Subunit V0C Regulates Aerobic Glycolysis of Esophageal Cancer Cells via PKM2 Signaling. Cells. 2019; 8(10):1137. https://doi.org/10.3390/cells8101137
Chicago/Turabian StyleSon, Sung Wook, Gia Cac Chau, Seong-Tae Kim, and Sung Hee Um. 2019. "Vacuolar H+-ATPase Subunit V0C Regulates Aerobic Glycolysis of Esophageal Cancer Cells via PKM2 Signaling" Cells 8, no. 10: 1137. https://doi.org/10.3390/cells8101137
APA StyleSon, S. W., Chau, G. C., Kim, S. -T., & Um, S. H. (2019). Vacuolar H+-ATPase Subunit V0C Regulates Aerobic Glycolysis of Esophageal Cancer Cells via PKM2 Signaling. Cells, 8(10), 1137. https://doi.org/10.3390/cells8101137