Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond



{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Pathophysiology of SLE at a Glance
1.2. Interaction between Environmental and Genetic Factors in SLE
1.3. The Roles of Germinal Centre and IgA Deficiency in SLE
1.4. Breach of Immune Tolerance in SLE
1.5. Contribution of Dysregulated Innate Immune System and Type-1 Interferon to SLE
2. Genetics in SLE
2.1. A Brief Background
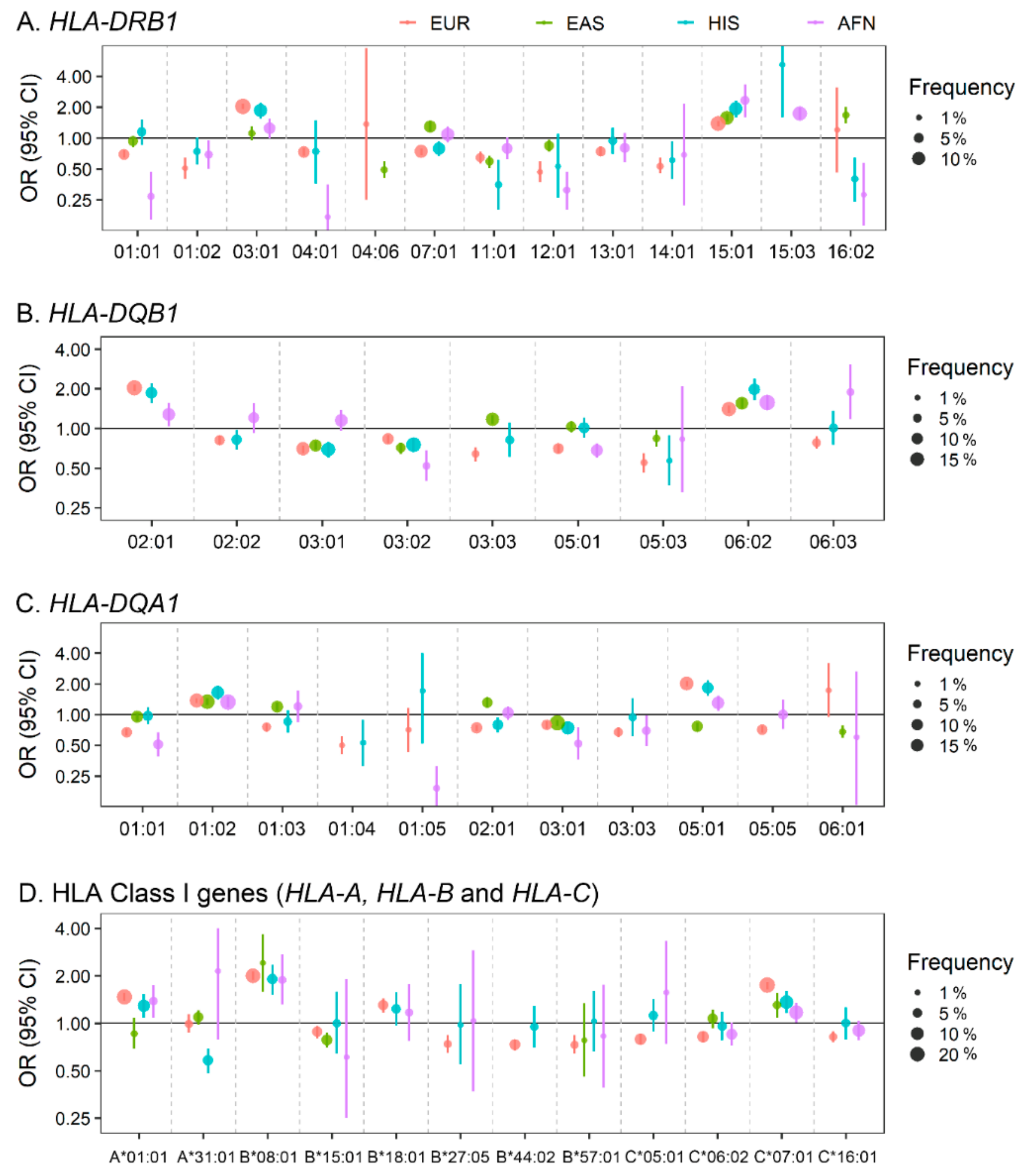
2.2. Classical and Non-Classical Human Leukocyte Antigen (HLA) Locus in SLE
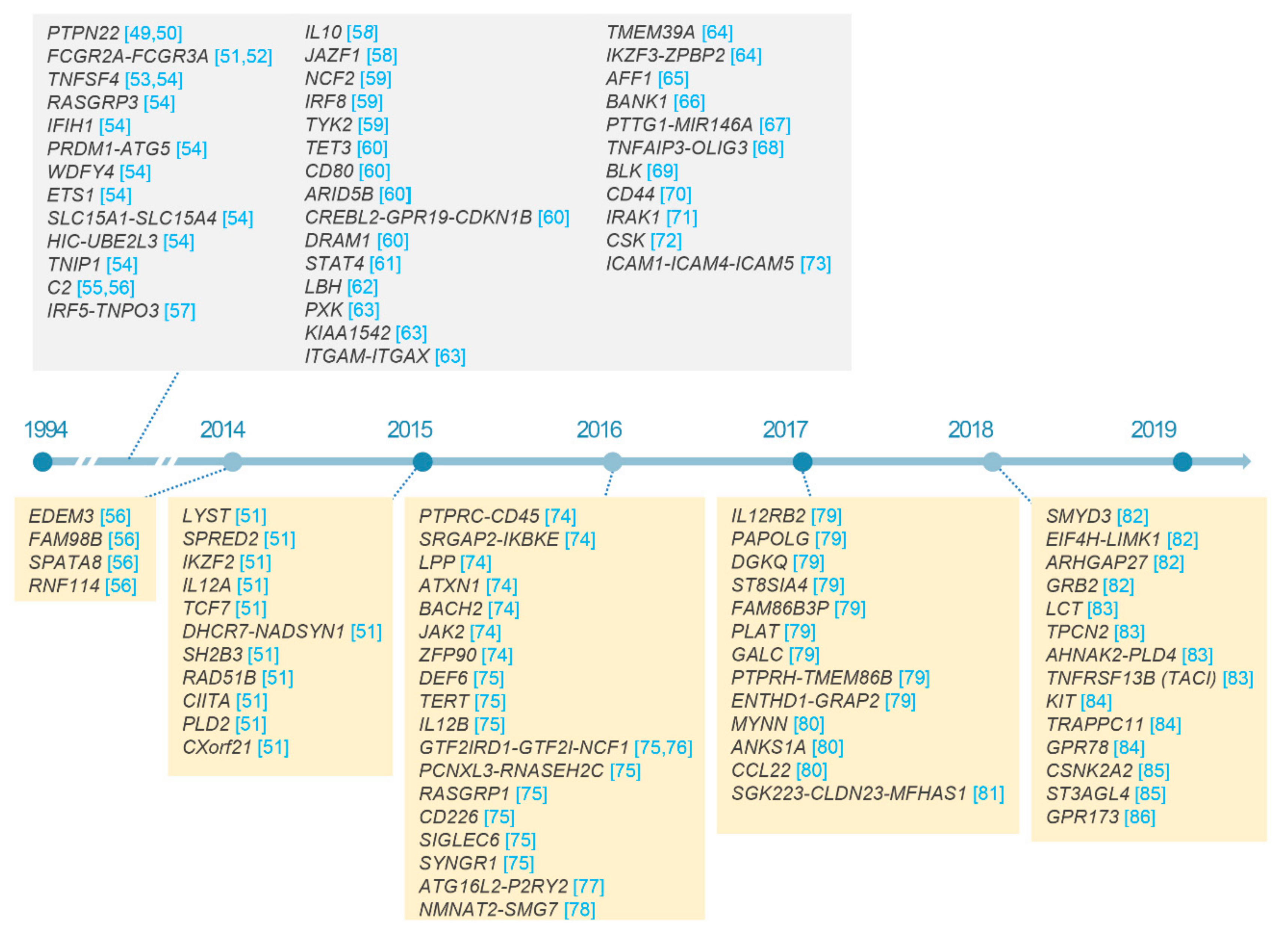
2.3. Non-HLA Loci in SLE
3. Immunobiological Insights from SLE-Risk Variants
4. Future Directions in Genetics of SLE and Conclusion
4.1. Integrative Analysis of Multi-Omics Data
4.2. Conclusions
Author Contributions
Conflicts of Interest
References
- Yacoub Wasef, S.Z. Gender differences in systemic lupus erythematosus. Gend. Med. 2004, 1, 12–17. [Google Scholar] [CrossRef]
- Lau, C.S.; Mak, A. The socioeconomic burden of SLE. Nat. Rev. Rheumatol. 2009, 5, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Mak, A.; Cheung, M.W.; Chiew, H.J.; Liu, Y.; Ho, R.C. Global trend of survival and damage of systemic lupus erythematosus: Meta-analysis and meta-regression of observational studies from the 1950s to 2000s. Semin. Arthritis Rheum. 2012, 41, 830–839. [Google Scholar] [CrossRef]
- Singh, R.R.; Yen, E.Y. SLE mortality remains disproportionately high, despite improvements over the last decade. Lupus 2018, 27, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.S.; Dooley, M.A.; Treadwell, E.L.; St Clair, E.W.; Parks, C.G.; Gilkeson, G.S. Hormonal, environmental, and infectious risk factors for developing systemic lupus erythematosus. Arthritis Rheum. 1998, 41, 1714–1724. [Google Scholar] [CrossRef]
- Mak, A.; Tay, S.H. Environmental factors, toxicants and systemic lupus erythematosus. Int. J. Mol. Sci. 2014, 15, 16043–16056. [Google Scholar] [CrossRef]
- Cooper, G.S.; Dooley, M.A.; Treadwell, E.L.; St Clair, E.W.; Gilkeson, G.S. Hormonal and reproductive risk factors for development of systemic lupus erythematosus: Results of a population-based, case-control study. Arthritis Rheum. 2002, 46, 1830–1839. [Google Scholar] [CrossRef]
- Khamashta, M.A. Systemic lupus erythematosus and pregnancy. Best Pract. Res. Clin. Rheumatol. 2006, 20, 685–694. [Google Scholar] [CrossRef]
- Imbasciati, E.; Tincani, A.; Gregorini, G.; Doria, A.; Moroni, G.; Cabiddu, G.; Marcelli, D. Pregnancy in women with pre-existing lupus nephritis: Predictors of fetal and maternal outcome. Nephrol. Dial. Transplant. 2009, 24, 519–525. [Google Scholar] [CrossRef]
- Buyon, J.P.; Petri, M.A.; Kim, M.Y.; Kalunian, K.C.; Grossman, J.; Hahn, B.H.; Merrill, J.T.; Sammaritano, L.; Lockshin, M.; Alarcon, G.S.; et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: A randomized trial. Ann. Intern. Med. 2005, 142, 953–962. [Google Scholar] [CrossRef]
- Kanda, N.; Tsuchida, T.; Tamaki, K. Estrogen enhancement of anti-double-stranded DNA antibody and immunoglobulin G production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 328–337. [Google Scholar] [CrossRef]
- Sthoeger, Z.M.; Chiorazzi, N.; Lahita, R.G. Regulation of the immune response by sex hormones. I. In vitro effects of estradiol and testosterone on pokeweed mitogen-induced human B cell differentiation. J. Immunol. 1988, 141, 91–98. [Google Scholar]
- Rider, V.; Jones, S.; Evans, M.; Bassiri, H.; Afsar, Z.; Abdou, N.I. Estrogen increases CD40 ligand expression in T cells from women with systemic lupus erythematosus. J. Rheumatol. 2001, 28, 2644–2649. [Google Scholar]
- Smith-Bouvier, D.L.; Divekar, A.A.; Sasidhar, M.; Du, S.; Tiwari-Woodruff, S.K.; King, J.K.; Arnold, A.P.; Singh, R.R.; Voskuhl, R.R. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 2008, 205, 1099–1108. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, C.M. Mechanistic aspects of epigenetic dysregulation in SLE. Clin. Immunol. 2018, 196, 3–11. [Google Scholar] [CrossRef]
- Coit, P.; Dozmorov, M.G.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Wren, J.D.; Sawalha, A.H. Epigenetic Reprogramming in Naive CD4+ T Cells Favoring T Cell Activation and Non-Th1 Effector T Cell Immune Response as an Early Event in Lupus Flares. Arthritis Rheumatol. 2016, 68, 2200–2209. [Google Scholar] [CrossRef]
- Laisne, M.; Gupta, N.; Kirsh, O.; Pradhan, S.; Defossez, P.A. Mechanisms of DNA Methyltransferase Recruitment in Mammals. Genes 2018, 9, 617. [Google Scholar] [CrossRef]
- Jeffries, M.A.; Dozmorov, M.; Tang, Y.; Merrill, J.T.; Wren, J.D.; Sawalha, A.H. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 2011, 6, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, H.; Liu, S.; Xia, F.; Kang, Z.; Zhang, Y.; Liu, Y.; Xiao, H.; Chen, L.; Huang, C.; et al. Excessive CD11c(+)Tbet(+) B cells promote aberrant TFH differentiation and affinity-based germinal center selection in lupus. Proc. Natl. Acad. Sci. USA 2019, 116, 18550–18560. [Google Scholar] [CrossRef]
- Zhao, L.D.; Liang, D.; Wu, X.N.; Li, Y.; Niu, J.W.; Zhou, C.; Wang, L.; Chen, H.; Zheng, W.J.; Fei, Y.Y.; et al. Contribution and underlying mechanisms of CXCR4 overexpression in patients with systemic lupus erythematosus. Cell. Mol. Immunol. 2017, 14, 842–849. [Google Scholar] [CrossRef]
- Wu, B.X.; Zhao, L.D.; Zhang, X. CXCR4 and CXCR5 orchestrate dynamic germinal center reactions and may contribute to the pathogenesis of systemic lupus erythematosus. Cell. Mol. Immunol. 2019, 16, 724–726. [Google Scholar] [CrossRef]
- Rankin, E.C.; Isenberg, D.A. IgA deficiency and SLE: Prevalence in a clinic population and a review of the literature. Lupus 1997, 6, 390–394. [Google Scholar]
- Cassidy, J.T.; Kitson, R.K.; Selby, C.L. Selective IgA deficiency in children and adults with systemic lupus erythematosus. Lupus 2007, 16, 647–650. [Google Scholar] [CrossRef]
- Latiff, A.H.; Kerr, M.A. The clinical significance of immunoglobulin A deficiency. Ann. Clin. Biochem. 2007, 44, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Rezvan, H.; Ahmadi, D.; Esmailzadeh, S.; Dayhimi, I. Selective deficiency of immunoglobulin A among healthy voluntary blood donors in Iran. Blood Transfus. 2009, 7, 152–154. [Google Scholar]
- Pieterse, E.; van der Vlag, J. Breaking immunological tolerance in systemic lupus erythematosus. Front. Immunol. 2014, 5, 164. [Google Scholar] [CrossRef]
- Reefman, E.; Horst, G.; Nijk, M.T.; Limburg, P.C.; Kallenberg, C.G.; Bijl, M. Opsonization of late apoptotic cells by systemic lupus erythematosus autoantibodies inhibits their uptake via an Fcgamma receptor-dependent mechanism. Arthritis Rheum. 2007, 56, 3399–3411. [Google Scholar] [CrossRef]
- Niederer, H.A.; Clatworthy, M.R.; Willcocks, L.C.; Smith, K.G. FcgammaRIIB, FcgammaRIIIB, and systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2010, 1183, 69–88. [Google Scholar] [CrossRef]
- Clatworthy, M.R.; Willcocks, L.; Urban, B.; Langhorne, J.; Williams, T.N.; Peshu, N.; Watkins, N.A.; Floto, R.A.; Smith, K.G. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proc. Natl. Acad. Sci. USA 2007, 104, 7169–7174. [Google Scholar] [CrossRef]
- Jhou, J.P.; Yu, I.S.; Hwai, H.; Chen, C.S.; Chen, P.L.; Tzeng, S.J. The Lupus-Associated Fcgamma Receptor IIb-I232T Polymorphism Results in Impairment in the Negative Selection of Low-Affinity Germinal Center B Cells Via c-Abl in Mice. Arthritis Rheumatol. 2018, 70, 1866–1878. [Google Scholar]
- Bekircan-Kurt, C.E.; Tuncer Kurne, A.; Erdem-Ozdamar, S.; Kalyoncu, U.; Karabudak, R.; Tan, E. The course of myasthenia gravis with systemic lupus erythematosus. Eur. Neurol. 2014, 72, 326–329. [Google Scholar] [CrossRef]
- Sawamura, S.; Kajihara, I.; Makino, K.; Makino, T.; Fukushima, S.; Jinnin, M.; Oyama, B.; Hashimoto, T.; Ihn, H. Systemic lupus erythematosus associated with myasthenia gravis, pemphigus foliaceus and chronic thyroiditis after thymectomy. Australas. J. Dermatol. 2017, 58, e120–e122. [Google Scholar] [CrossRef]
- Miskovic, R.; Plavsic, A.; Peric-Popadic, A.; Raskovic, S.; Bogic, M. Systemic Lupus Erythematosus and Secondary Antiphospholipid Syndrome after Thymectomy for Myasthenia Gravis—A Case Report. Open Access Maced. J. Med. Sci. 2015, 3, 439–442. [Google Scholar] [CrossRef]
- Liu, R.; Dar, A.R.; Tay, K.Y.; Nicolle, M.W.; Inculet, R.I. Thymoma-associated Myasthenia Gravis in a Young Adult with Development of Paraneoplastic Limbic Encephalitis and Systemic Lupus Erythematosus Post-thymectomy: A Case Report. Cureus 2018, 10, e3581. [Google Scholar] [CrossRef] [Green Version]
- Murayama, G.; Furusawa, N.; Chiba, A.; Yamaji, K.; Tamura, N.; Miyake, S. Enhanced IFN-alpha production is associated with increased TLR7 retention in the lysosomes of palasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2017, 19, 234. [Google Scholar] [CrossRef]
- Sakata, K.; Nakayamada, S.; Miyazaki, Y.; Kubo, S.; Ishii, A.; Nakano, K.; Tanaka, Y. Up-Regulation of TLR7-Mediated IFN-alpha Production by Plasmacytoid Dendritic Cells in Patients With Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1957. [Google Scholar] [CrossRef]
- Chiche, L.; Jourde-Chiche, N.; Whalen, E.; Presnell, S.; Gersuk, V.; Dang, K.; Anguiano, E.; Quinn, C.; Burtey, S.; Berland, Y.; et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014, 66, 1583–1595. [Google Scholar] [CrossRef]
- Banchereau, J.; Pascual, V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef]
- Han, G.M.; Chen, S.L.; Shen, N.; Ye, S.; Bao, C.D.; Gu, Y.Y. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun. 2003, 4, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.Z.; Zhou, J.; Lian, Y.; Zhang, B.; Branch, V.K.; Carr-Johnson, F.; Karp, D.R.; Mohan, C.; Wakeland, E.K.; Olsen, N.J. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clin. Exp. Immunol. 2010, 159, 281–291. [Google Scholar] [CrossRef]
- Kariuki, S.N.; Ghodke-Puranik, Y.; Dorschner, J.M.; Chrabot, B.S.; Kelly, J.A.; Tsao, B.P.; Kimberly, R.P.; Alarcon-Riquelme, M.E.; Jacob, C.O.; Criswell, L.A.; et al. Genetic analysis of the pathogenic molecular sub-phenotype interferon-alpha identifies multiple novel loci involved in systemic lupus erythematosus. Genes Immun. 2015, 16, 15–23. [Google Scholar]
- Ko, K.; Koldobskaya, Y.; Rosenzweig, E.; Niewold, T.B. Activation of the Interferon Pathway is Dependent Upon Autoantibodies in African-American SLE Patients, but Not in European-American SLE Patients. Front. Immunol. 2013, 4, 309. [Google Scholar] [Green Version]
- Xue, Z.; Cui, C.; Liao, Z.; Xia, S.; Zhang, P.; Qin, J.; Guo, Q.; Chen, S.; Fu, Q.; Yin, Z.; et al. Identification of LncRNA Linc00513 Containing Lupus-Associated Genetic Variants as a Novel Regulator of Interferon Signaling Pathway. Front. Immunol. 2018, 9, 2967. [Google Scholar] [CrossRef]
- Kalunian, K.C.; Merrill, J.T.; Maciuca, R.; McBride, J.M.; Townsend, M.J.; Wei, X.; Davis, J.C., Jr.; Kennedy, W.P. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann. Rheum. Dis. 2016, 75, 196–202. [Google Scholar]
- Lawrence, J.S.; Martins, C.L.; Drake, G.L. A family survey of lupus erythematosus. 1. Heritability. J. Rheumatol. 1987, 14, 913–921. [Google Scholar]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar]
- Marchini, J.; Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010, 11, 499–511. [Google Scholar] [CrossRef]
- Kyogoku, C.; Langefeld, C.D.; Ortmann, W.A.; Lee, A.; Selby, S.; Carlton, V.E.; Chang, M.; Ramos, P.; Baechler, E.C.; Batliwalla, F.M.; et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am. J. Hum. Genet. 2004, 75, 504–507. [Google Scholar] [CrossRef]
- Namjou, B.; Kim-Howard, X.; Sun, C.; Adler, A.; Chung, S.A.; Kaufman, K.M.; Kelly, J.A.; Glenn, S.B.; Guthridge, J.M.; Scofield, R.H.; et al. PTPN22 association in systemic lupus erythematosus (SLE) with respect to individual ancestry and clinical sub-phenotypes. PLoS ONE 2013, 8, e69404. [Google Scholar] [CrossRef]
- Bentham, J.; Morris, D.L.; Graham, D.S.C.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martin, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef]
- Edberg, J.C.; Langefeld, C.D.; Wu, J.; Moser, K.L.; Kaufman, K.M.; Kelly, J.; Bansal, V.; Brown, W.M.; Salmon, J.E.; Rich, S.S.; et al. Genetic linkage and association of Fcgamma receptor IIIA (CD16A) on chromosome 1q23 with human systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 2132–2140. [Google Scholar] [CrossRef]
- Cunninghame Graham, D.S.; Graham, R.R.; Manku, H.; Wong, A.K.; Whittaker, J.C.; Gaffney, P.M.; Moser, K.L.; Rioux, J.D.; Altshuler, D.; Behrens, T.W.; et al. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat. Genet. 2008, 40, 83–89. [Google Scholar] [CrossRef]
- Han, J.W.; Zheng, H.F.; Cui, Y.; Sun, L.D.; Ye, D.Q.; Hu, Z.; Xu, J.H.; Cai, Z.M.; Huang, W.; Zhao, G.P.; et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1234–1237. [Google Scholar] [CrossRef]
- Sullivan, K.E.; Petri, M.A.; Schmeckpeper, B.J.; McLean, R.H.; Winkelstein, J.A. Prevalence of a mutation causing C2 deficiency in systemic lupus erythematosus. J. Rheumatol. 1994, 21, 1128–1133. [Google Scholar]
- Armstrong, D.L.; Zidovetzki, R.; Alarcon-Riquelme, M.E.; Tsao, B.P.; Criswell, L.A.; Kimberly, R.P.; Harley, J.B.; Sivils, K.L.; Vyse, T.J.; Gaffney, P.M.; et al. GWAS identifies novel SLE susceptibility genes and explains the association of the HLA region. Genes Immun. 2014, 15, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.R.; Kozyrev, S.V.; Baechler, E.C.; Reddy, M.V.; Plenge, R.M.; Bauer, J.W.; Ortmann, W.A.; Koeuth, T.; Gonzalez Escribano, M.F.; Argentine and Spanish Collaborative Groups; et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat. Genet. 2006, 38, 550–555. [Google Scholar] [CrossRef]
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233. [Google Scholar] [CrossRef] [Green Version]
- Cunninghame Graham, D.S.; Morris, D.L.; Bhangale, T.R.; Criswell, L.A.; Syvanen, A.C.; Ronnblom, L.; Behrens, T.W.; Graham, R.R.; Vyse, T.J. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 2011, 7, e1002341. [Google Scholar] [CrossRef]
- Yang, W.; Tang, H.; Zhang, Y.; Tang, X.; Zhang, J.; Sun, L.; Yang, J.; Cui, Y.; Zhang, L.; Hirankarn, N.; et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am. J. Hum. Genet. 2013, 92, 41–51. [Google Scholar] [CrossRef]
- Remmers, E.F.; Plenge, R.M.; Lee, A.T.; Graham, R.R.; Hom, G.; Behrens, T.W.; de Bakker, P.I.; Le, J.M.; Lee, H.S.; Batliwalla, F.; et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl. J. Med. 2007, 357, 977–986. [Google Scholar] [CrossRef]
- Yu, Z.Y.; Lu, W.S.; Zuo, X.B.; Hu, J.; Yao, S.; Li, Y.; Han, J.W.; Sun, L.D.; Cheng, Y.L.; Xu, Q.; et al. One novel susceptibility locus associate with systemic lupus erythematosus in Chinese Han population. Rheumatol. Int. 2013, 33, 2079–2083. [Google Scholar] [CrossRef]
- International Consortium for Systemic Lupus Erythematosus Genetics; Harley, J.B.; Alarcon-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D.; et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008, 40, 204–210. [Google Scholar] [CrossRef]
- Lessard, C.J.; Adrianto, I.; Ice, J.A.; Wiley, G.B.; Kelly, J.A.; Glenn, S.B.; Adler, A.J.; Li, H.; Rasmussen, A.; Williams, A.H.; et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am. J. Hum. Genet. 2012, 90, 648–660. [Google Scholar] [CrossRef]
- Okada, Y.; Shimane, K.; Kochi, Y.; Tahira, T.; Suzuki, A.; Higasa, K.; Takahashi, A.; Horita, T.; Atsumi, T.; Ishii, T.; et al. A genome-wide association study identified AFF1 as a susceptibility locus for systemic lupus eyrthematosus in Japanese. PLoS Genet. 2012, 8, e1002455. [Google Scholar] [CrossRef]
- Kozyrev, S.V.; Abelson, A.K.; Wojcik, J.; Zaghlool, A.; Linga Reddy, M.V.; Sanchez, E.; Gunnarsson, I.; Svenungsson, E.; Sturfelt, G.; Jonsen, A.; et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 211–216. [Google Scholar] [CrossRef]
- Luo, X.; Yang, W.; Ye, D.Q.; Cui, H.; Zhang, Y.; Hirankarn, N.; Qian, X.; Tang, Y.; Lau, Y.L.; de Vries, N.; et al. A functional variant in microRNA-146a promoter modulates its expression and confers disease risk for systemic lupus erythematosus. PLoS Genet. 2011, 7, e1002128. [Google Scholar] [CrossRef]
- Graham, R.R.; Cotsapas, C.; Davies, L.; Hackett, R.; Lessard, C.J.; Leon, J.M.; Burtt, N.P.; Guiducci, C.; Parkin, M.; Gates, C.; et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1059–1061. [Google Scholar] [CrossRef]
- Hom, G.; Graham, R.R.; Modrek, B.; Taylor, K.E.; Ortmann, W.; Garnier, S.; Lee, A.T.; Chung, S.A.; Ferreira, R.C.; Pant, P.V.; et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 2008, 358, 900–909. [Google Scholar] [CrossRef]
- Lessard, C.J.; Adrianto, I.; Kelly, J.A.; Kaufman, K.M.; Grundahl, K.M.; Adler, A.; Williams, A.H.; Gallant, C.J.; Marta, E.A.-R.o.b.o.t.B.; Networks, G.; et al. Identification of a systemic lupus erythematosus susceptibility locus at 11p13 between PDHX and CD44 in a multiethnic study. Am. J. Hum. Genet. 2011, 88, 83–91. [Google Scholar] [CrossRef]
- Jacob, C.O.; Zhu, J.; Armstrong, D.L.; Yan, M.; Han, J.; Zhou, X.J.; Thomas, J.A.; Reiff, A.; Myones, B.L.; Ojwang, J.O.; et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2009, 106, 6256–6261. [Google Scholar] [CrossRef] [PubMed]
- Manjarrez-Orduno, N.; Marasco, E.; Chung, S.A.; Katz, M.S.; Kiridly, J.F.; Simpfendorfer, K.R.; Freudenberg, J.; Ballard, D.H.; Nashi, E.; Hopkins, T.J.; et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat. Genet. 2012, 44, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Brown, E.E.; Choi, C.B.; Alarcon-Riquelme, M.E.; Biolupus; Kelly, J.A.; Glenn, S.B.; Ojwang, J.O.; Adler, A.; Lee, H.S.; et al. Variation in the ICAM1-ICAM4-ICAM5 locus is associated with systemic lupus erythematosus susceptibility in multiple ancestries. Ann. Rheum. Dis. 2012, 71, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Sheng, Y.; Zhang, Y.; Wang, Y.F.; Zhu, Z.; Tombleson, P.; Chen, L.; Cunninghame Graham, D.S.; Bentham, J.; Roberts, A.L.; et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 2016, 48, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Molineros, J.E.; Looger, L.L.; Zhou, X.J.; Kim, K.; Okada, Y.; Ma, J.; Qi, Y.Y.; Kim-Howard, X.; Motghare, P.; et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat. Genet. 2016, 48, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Ma, J.; Deng, Y.; Kelly, J.A.; Kim, K.; Bang, S.Y.; Lee, H.S.; Li, Q.Z.; Wakeland, E.K.; Qiu, R.; et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat. Genet. 2017, 49, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessard, C.J.; Sajuthi, S.; Zhao, J.; Kim, K.; Ice, J.A.; Li, H.; Ainsworth, H.; Rasmussen, A.; Kelly, J.A.; Marion, M.; et al. Identification of a Systemic Lupus Erythematosus Risk Locus Spanning ATG16L2, FCHSD2, and P2RY2 in Koreans. Arthritis Rheumatol. 2016, 68, 1197–1209. [Google Scholar] [PubMed]
- Deng, Y.; Zhao, J.; Sakurai, D.; Sestak, A.L.; Osadchiy, V.; Langefeld, C.D.; Kaufman, K.M.; Kelly, J.A.; James, J.A.; Petri, M.A.; et al. Decreased SMG7 expression associates with lupus-risk variants and elevated antinuclear antibody production. Ann. Rheum. Dis. 2016, 75, 2007–2013. [Google Scholar] [CrossRef] [Green Version]
- Langefeld, C.D.; Ainsworth, H.C.; Cunninghame Graham, D.S.; Kelly, J.A.; Comeau, M.E.; Marion, M.C.; Howard, T.D.; Ramos, P.S.; Croker, J.A.; Morris, D.L.; et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 2017, 8, 16021. [Google Scholar] [CrossRef]
- Molineros, J.E.; Yang, W.; Zhou, X.J.; Sun, C.; Okada, Y.; Zhang, H.; Heng Chua, K.; Lau, Y.L.; Kochi, Y.; Suzuki, A.; et al. Confirmation of five novel susceptibility loci for systemic lupus erythematosus (SLE) and integrated network analysis of 82 SLE susceptibility loci. Hum. Mol. Genet. 2017, 26, 1205–1216. [Google Scholar] [CrossRef]
- Demirci, F.Y.; Wang, X.; Morris, D.L.; Feingold, E.; Bernatsky, S.; Pineau, C.; Clarke, A.; Ramsey-Goldman, R.; Manzi, S.; Vyse, T.J.; et al. Multiple signals at the extended 8p23 locus are associated with susceptibility to systemic lupus erythematosus. J. Med. Genet. 2017, 54, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Julia, A.; Lopez-Longo, F.J.; Perez Venegas, J.J.; Bonas-Guarch, S.; Olive, A.; Andreu, J.L.; Aguirre-Zamorano, M.A.; Vela, P.; Nolla, J.M.; de la Fuente, J.L.M.; et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Zhu, C.; Zhu, Z.; Yang, C.; Zheng, X.; Liu, L.; Zuo, X.; Sheng, Y.; Tang, H.; Liang, B.; et al. Exome-wide association study identifies four novel loci for systemic lupus erythematosus in Han Chinese population. Ann. Rheum. Dis. 2018, 77, 417. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zuo, X.; Zhu, Z.; Wen, L.; Yang, C.; Zhu, C.; Tang, L.; Cheng, Y.; Chen, M.; Zhou, F.; et al. Genome-wide association study identifies three novel susceptibility loci for systemic lupus erythematosus in Han Chinese. Br. J. Dermatol. 2018, 179, 506–508. [Google Scholar] [PubMed]
- Wang, Y.F.; Zhang, Y.; Zhu, Z.; Wang, T.Y.; Morris, D.L.; Shen, J.J.; Zhang, H.; Pan, H.F.; Yang, J.; Yang, S.; et al. Identification of ST3AGL4, MFHAS1, CSNK2A2 and CD226 as loci associated with systemic lupus erythematosus (SLE) and evaluation of SLE genetics in drug repositioning. Ann. Rheum. Dis. 2018, 77, 1078–1084. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.; Wang, Y.F.; Morris, D.; Hirankarn, N.; Sheng, Y.; Shen, J.; Pan, H.F.; Yang, J.; Yang, S.; et al. Meta-analysis of GWAS on both Chinese and European populations identifies GPR173 as a novel X chromosome susceptibility gene for SLE. Arthritis Res. Ther. 2018, 20, 92. [Google Scholar] [CrossRef] [Green Version]
- Trowsdale, J.; Knight, J.C. Major histocompatibility complex genomics and human disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.; Wilming, L.; Rand, V.; Lovering, R.C.; Bruford, E.A.; Khodiyar, V.K.; Lush, M.J.; Povey, S.; Talbot, C.C., Jr.; Wright, M.W.; et al. Gene map of the extended human MHC. Nat. Rev. Genet. 2004, 5, 889–899. [Google Scholar] [CrossRef]
- Jia, X.; Han, B.; Onengut-Gumuscu, S.; Chen, W.M.; Concannon, P.J.; Rich, S.S.; Raychaudhuri, S.; de Bakker, P.I. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE 2013, 8, e64683. [Google Scholar] [CrossRef]
- Dilthey, A.T.; Moutsianas, L.; Leslie, S.; McVean, G. HLA*IMP—An integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics 2011, 27, 968–972. [Google Scholar] [CrossRef]
- Lim, J.; Bae, S.C.; Kim, K. Understanding HLA associations from SNP summary association statistics. Sci. Rep. 2019, 9, 1337. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.Y.; Choi, J.Y.; Park, S.; Choi, J.; Hong, S.J.; Lee, H.S.; Choi, C.B.; Bae, S.C. Brief Report: Influence of HLA-DRB1 Susceptibility Alleles on the Clinical Subphenotypes of Systemic Lupus Erythematosus in Koreans. Arthritis Rheumatol. 2016, 68, 1190–1196. [Google Scholar] [PubMed]
- Morris, D.L.; Taylor, K.E.; Fernando, M.M.; Nititham, J.; Alarcon-Riquelme, M.E.; Barcellos, L.F.; Behrens, T.W.; Cotsapas, C.; Gaffney, P.M.; Graham, R.R.; et al. Unraveling multiple MHC gene associations with systemic lupus erythematosus: Model choice indicates a role for HLA alleles and non-HLA genes in Europeans. Am. J. Hum. Genet. 2012, 91, 778–793. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Bang, S.Y.; Lee, H.S.; Okada, Y.; Han, B.; Saw, W.Y.; Teo, Y.Y.; Bae, S.C. The HLA-DRbeta1 amino acid positions 11-13-26 explain the majority of SLE-MHC associations. Nat. Commun. 2014, 5, 5902. [Google Scholar] [CrossRef] [PubMed]
- Molineros, J.E.; Looger, L.L.; Kim, K.; Okada, Y.; Terao, C.; Sun, C.; Zhou, X.J.; Raj, P.; Kochi, Y.; Suzuki, A.; et al. Amino acid signatures of HLA Class-I and II molecules are strongly associated with SLE susceptibility and autoantibody production in Eastern Asians. PLoS Genet. 2019, 15, e1008092. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Ohmura, K.; Yamada, R.; Kawaguchi, T.; Shimizu, M.; Tabara, Y.; Takahashi, M.; Setoh, K.; Nakayama, T.; Kosugi, S.; et al. Association between antinuclear antibodies and the HLA class II locus and heterogeneous characteristics of staining patterns: The Nagahama study. Arthritis Rheumatol. 2014, 66, 3395–3403. [Google Scholar] [CrossRef]
- Xu, R.; Li, Q.; Liu, R.; Shen, J.; Li, M.; Zhao, M.; Wang, M.; Liao, Q.; Mao, H.; Li, Z.; et al. Association Analysis of the MHC in Lupus Nephritis. J. Am. Soc. Nephrol. 2017, 28, 3383–3394. [Google Scholar] [CrossRef]
- Hachiya, Y.; Kawasaki, A.; Oka, S.; Kondo, Y.; Ito, S.; Matsumoto, I.; Kusaoi, M.; Amano, H.; Suda, A.; Setoguchi, K.; et al. Association of HLA-G 3′ Untranslated Region Polymorphisms with Systemic Lupus Erythematosus in a Japanese Population: A Case-Control Association Study. PLoS ONE 2016, 11, e0158065. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wu, Y.L.; Mok, M.Y.; Wu, Y.J.; Lintner, K.E.; Wang, C.M.; Chung, E.K.; Yang, Y.; Zhou, B.; Wang, H.; et al. Effects of Complement C4 Gene Copy Number Variations, Size Dichotomy, and C4A Deficiency on Genetic Risk and Clinical Presentation of Systemic Lupus Erythematosus in East Asian Populations. Arthritis Rheumatol. 2016, 68, 1442–1453. [Google Scholar] [CrossRef]
- Olsson, L.M.; Nerstedt, A.; Lindqvist, A.K.; Johansson, S.C.; Medstrand, P.; Olofsson, P.; Holmdahl, R. Copy number variation of the gene NCF1 is associated with rheumatoid arthritis. Antioxid. Redox Signal. 2012, 16, 71–78. [Google Scholar] [CrossRef]
- Ago, T.; Kuribayashi, F.; Hiroaki, H.; Takeya, R.; Ito, T.; Kohda, D.; Sumimoto, H. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc. Natl. Acad. Sci. USA 2003, 100, 4474–4479. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.B.; Lim, J.; Tsao, B.P.; Nath, S.K.; Kim, K.; Bae, S.C. Genetic variants in systemic lupus erythematosus susceptibility loci, XKR6 and GLT1D1 are associated with childhood-onset SLE in a Korean cohort. Sci. Rep. 2018, 8, 9962. [Google Scholar] [CrossRef]
- Taylor, K.E.; Chung, S.A.; Graham, R.R.; Ortmann, W.A.; Lee, A.T.; Langefeld, C.D.; Jacob, C.O.; Kamboh, M.I.; Alarcon-Riquelme, M.E.; Tsao, B.P.; et al. Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. PLoS Genet. 2011, 7, e1001311. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.; Kelly, J.A.; Somers, E.C.; Hughes, T.; Kaufman, K.M.; Sanchez, E.; Nath, S.K.; Bruner, G.; Alarcon-Riquelme, M.E.; Gilkeson, G.S.; et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann. Rheum. Dis. 2011, 70, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Trynka, G.; Sandor, C.; Han, B.; Xu, H.; Stranger, B.E.; Liu, X.S.; Raychaudhuri, S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 2013, 45, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shi, L.; Song, L.; Maurer, K.; Petri, M.A.; Sullivan, K.E. Overall Downregulation of mRNAs and Enrichment of H3K4me3 Change Near Genome-Wide Association Study Signals in Systemic Lupus Erythematosus: Cell-Specific Effects. Front. Immunol. 2018, 9, 497. [Google Scholar] [CrossRef]
- Lim, J.; Kim, K. Genetic variants differentially associated with rheumatoid arthritis and systemic lupus erythematosus reveal the disease-specific biology. Sci. Rep. 2019, 9, 2739. [Google Scholar] [CrossRef]
- Timmons, J.A.; Szkop, K.J.; Gallagher, I.J. Multiple sources of bias confound functional enrichment analysis of global-omics data. Genome Biol. 2015, 16, 186. [Google Scholar] [CrossRef]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165, 1548–1550. [Google Scholar] [CrossRef]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiers, M.; Minnoye, L.; Aibar, S.; Bravo Gonzalez-Blas, C.; Kalender Atak, Z.; Aerts, S. Mapping gene regulatory networks from single-cell omics data. Brief. Funct. Genom. 2018, 17, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Arazi, A.; Rao, D.A.; Berthier, C.C.; Davidson, A.; Liu, Y.; Hoover, P.J.; Chicoine, A.; Eisenhaure, T.M.; Jonsson, A.H.; Li, S.; et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 2019, 20, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Der, E.; Suryawanshi, H.; Morozov, P.; Kustagi, M.; Goilav, B.; Ranabathou, S.; Izmirly, P.; Clancy, R.; Belmont, H.M.; Koenigsberg, M.; et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol. 2019, 20, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.A.; Wegmann, D.; Trynka, G.; Gutierrez-Achury, J.; Do, R.; Voight, B.F.; Kraft, P.; Chen, R.; Kallberg, H.J.; Kurreeman, F.A.; et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat. Genet. 2012, 44, 483–489. [Google Scholar] [CrossRef]
- Fang, H.; Consortium, U.-D.; De Wolf, H.; Knezevic, B.; Burnham, K.L.; Osgood, J.; Sanniti, A.; Lledo Lara, A.; Kasela, S.; De Cesco, S.; et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 2019, 51, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M. Priority index for human genetics and drug discovery. Nat. Genet. 2019, 51, 1073–1075. [Google Scholar] [CrossRef]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 2013, 12, 581–594. [Google Scholar] [CrossRef]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, Y.-C.; Chun, S.; Kim, K.; Mak, A. Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond. Cells 2019, 8, 1180. https://doi.org/10.3390/cells8101180
Kwon Y-C, Chun S, Kim K, Mak A. Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond. Cells. 2019; 8(10):1180. https://doi.org/10.3390/cells8101180
Chicago/Turabian StyleKwon, Young-Chang, Sehwan Chun, Kwangwoo Kim, and Anselm Mak. 2019. "Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond" Cells 8, no. 10: 1180. https://doi.org/10.3390/cells8101180
APA StyleKwon, Y. -C., Chun, S., Kim, K., & Mak, A. (2019). Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond. Cells, 8(10), 1180. https://doi.org/10.3390/cells8101180