Clonal Evolution of TP53 c.375+1G>A Mutation in Pre- and Post- Neo-Adjuvant Chemotherapy (NACT) Tumor Samples in High-Grade Serous Ovarian Cancer (HGSOC)

 ,
,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Human Ethics
2.2. Next Generation Sequencing Analysis and Somatic LOH Analysis
2.3. Sanger Sequencing to Validate TP53 SNVs in Trans Configuration
2.4. Immunohistochemistry
3. Results
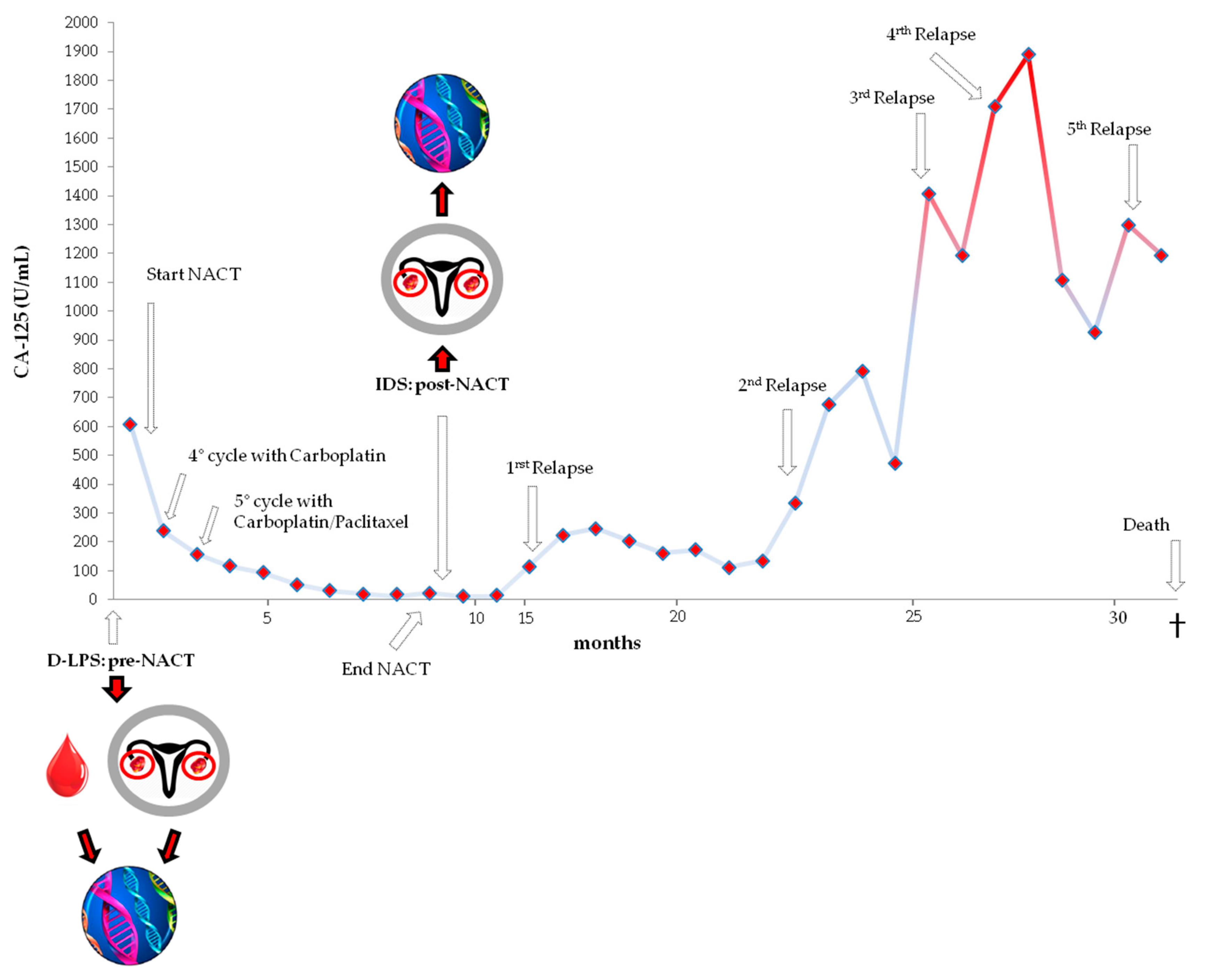
3.1. Case History
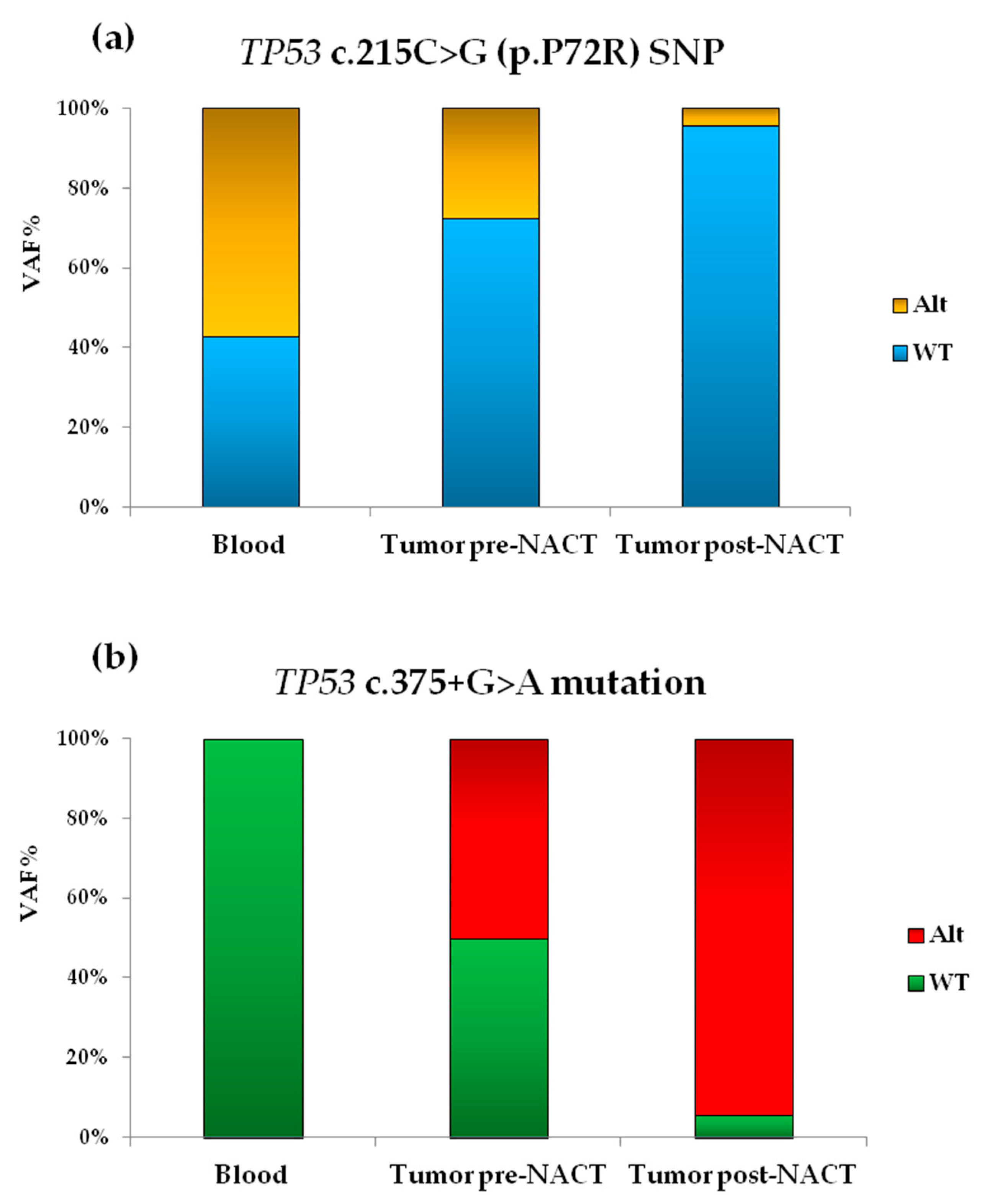
3.2. Molecular Analysis by NGS
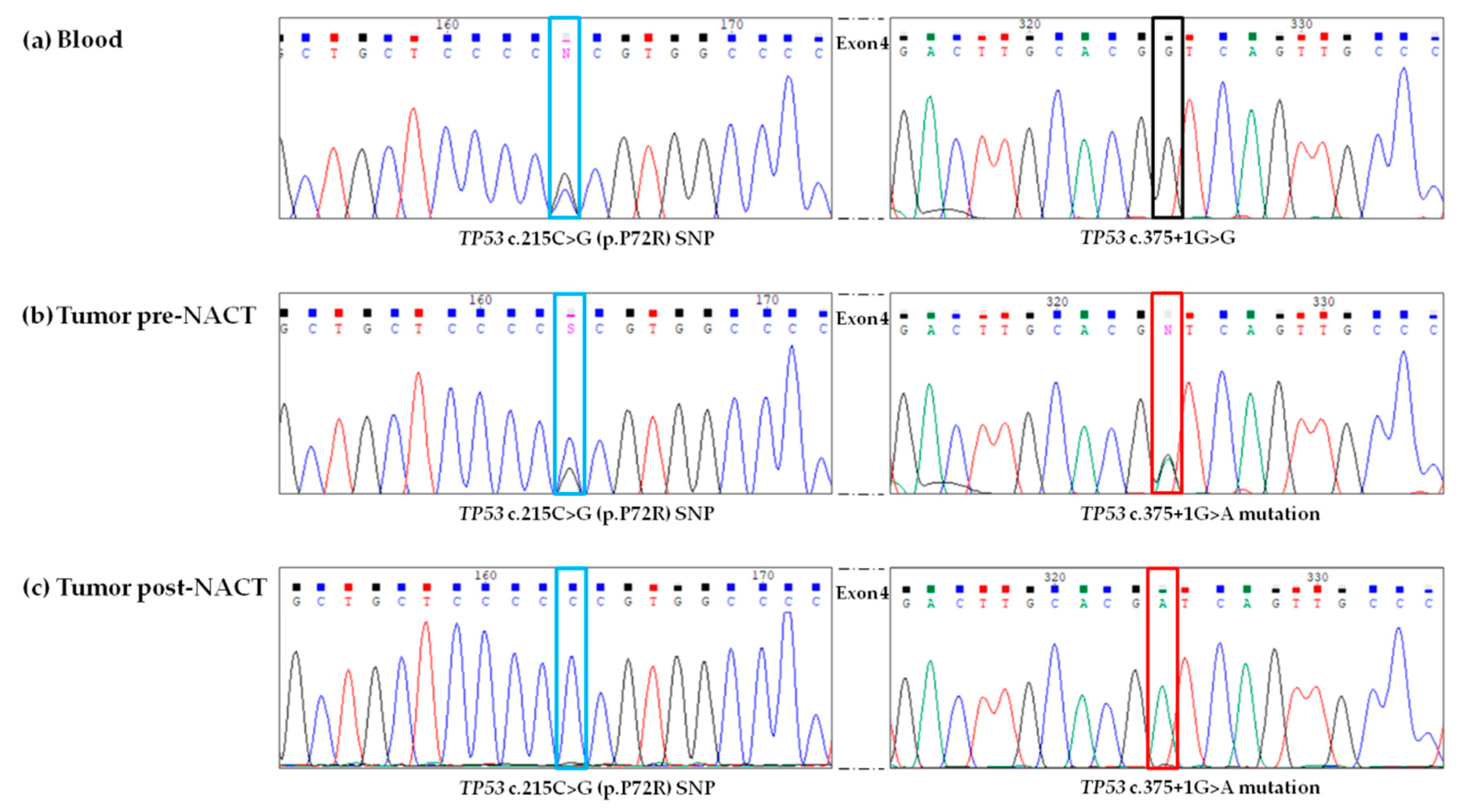
3.3. Validation of NGS Results by Sanger Sequencing
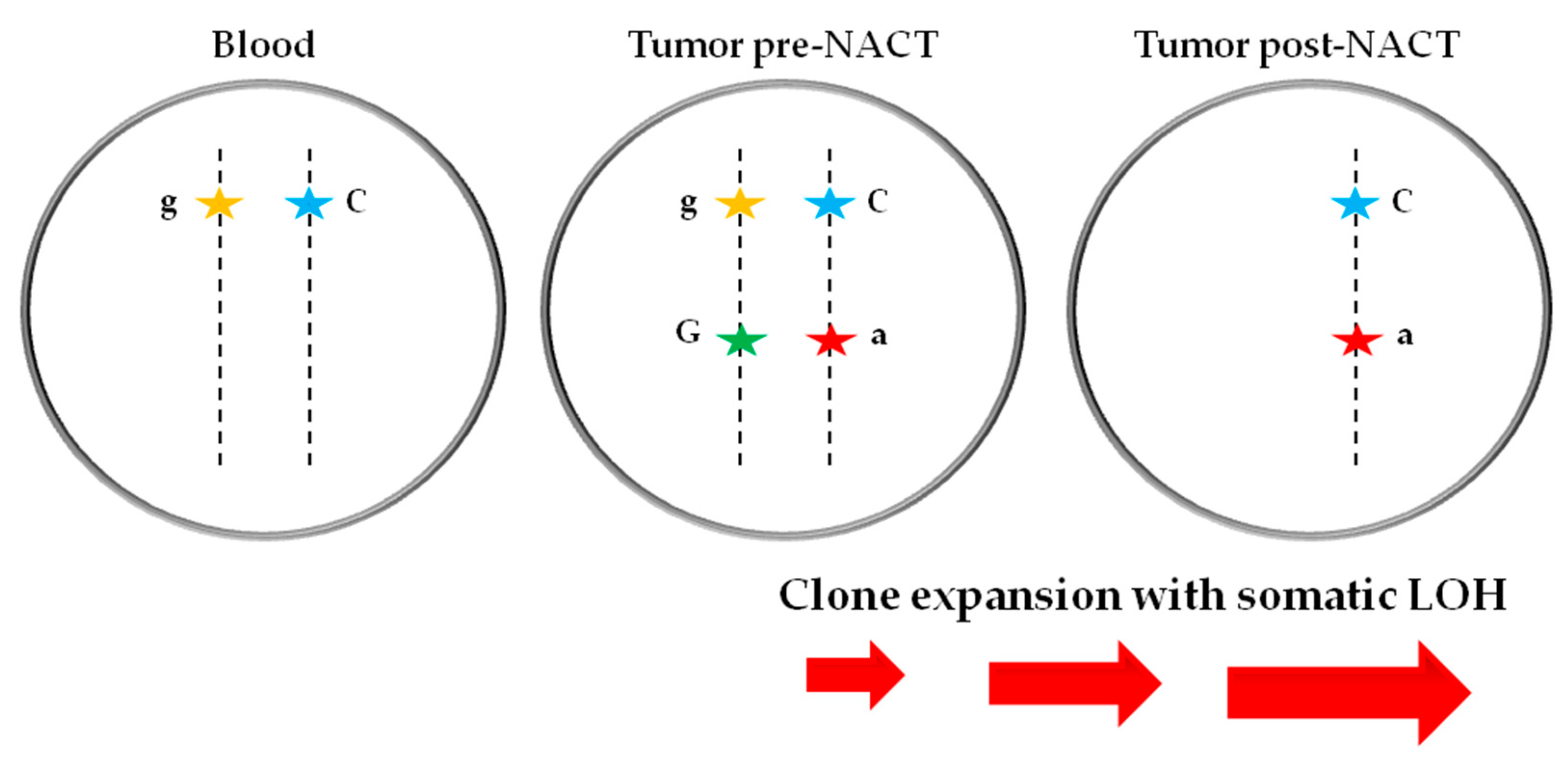
3.4. Somatic LOH
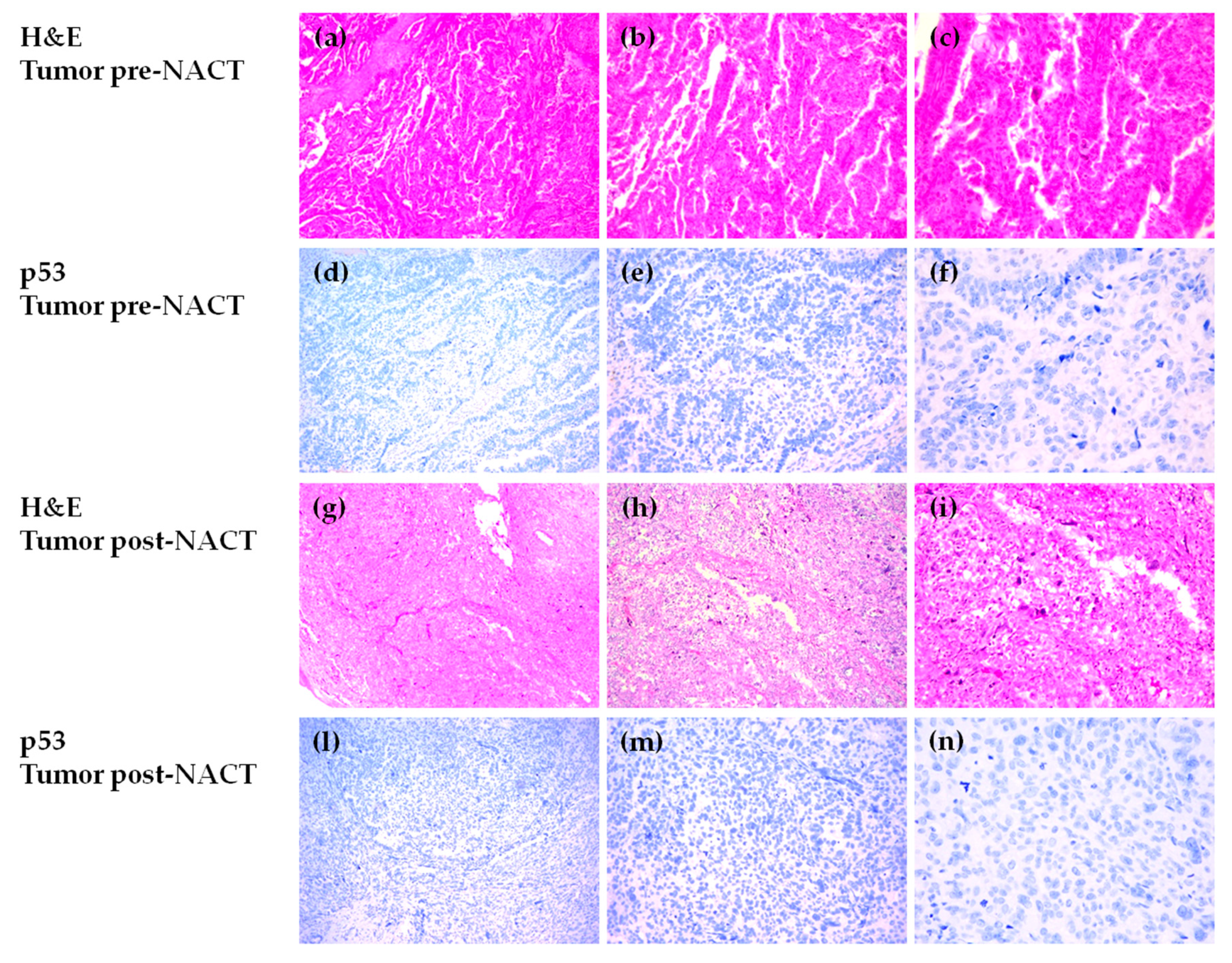
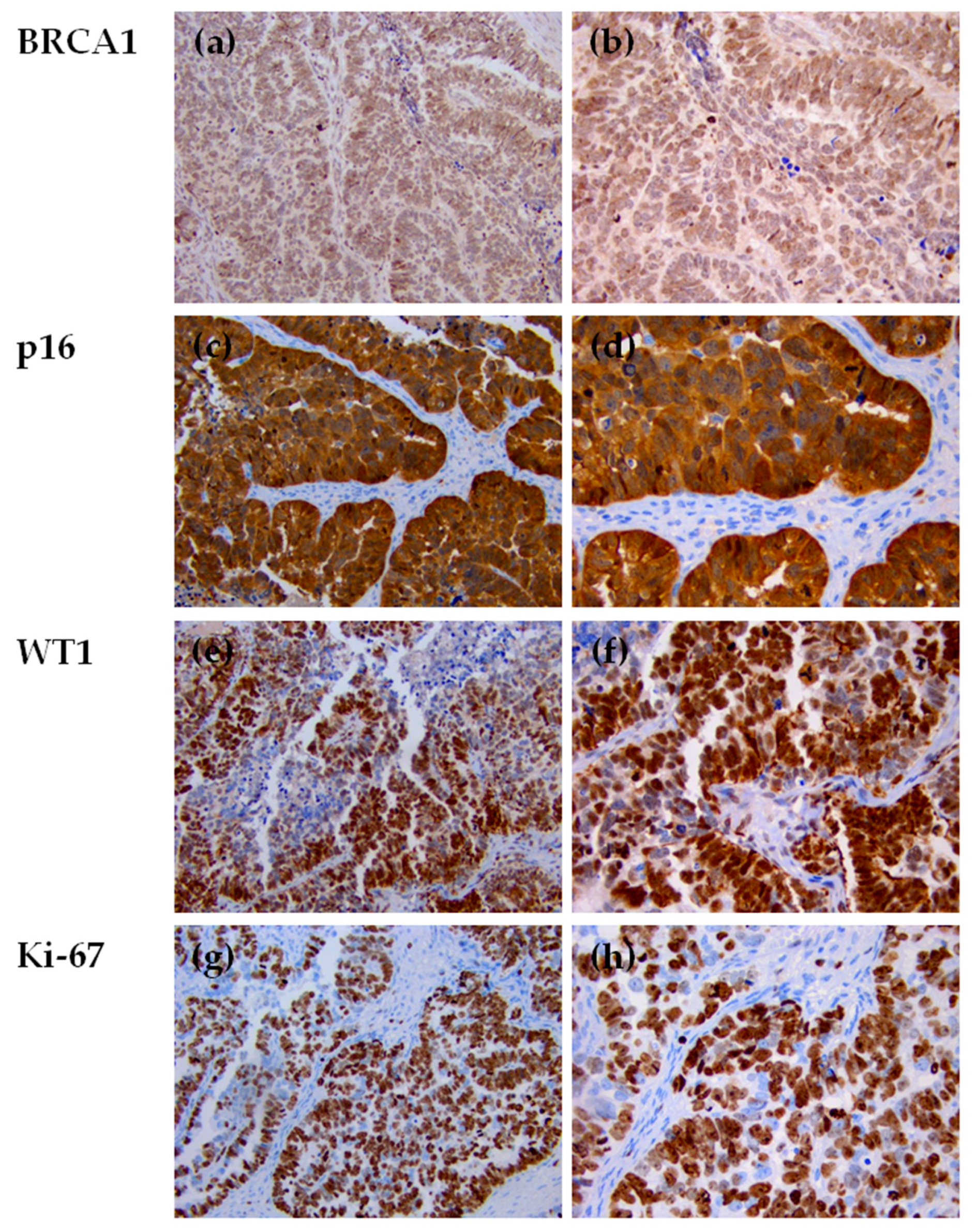
3.5. Immunohistochemical Evaluation of p53 Expression in HGSOC Tumor Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeyer, W.R.; Beach, J.A.; Karlan, B.Y. Reversing Platinum Resistance in High-Grade Serous Ovarian Carcinoma: Targeting BRCA and the Homologous Recombination System. Front. Oncol. 2014, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Associazione Italiana di Oncologia Medica (AIOM). Linee Guida Tumori dell’Ovaio 2018; AIOM: Italy, 2018; Available online: http://www.aiom.it/linee-guida-aiom-2018-tumori-dellovaio/ (accessed on 12 August 2019).
- Winter, W.E.; Maxwell, G.L.; Tian, C.; Carlson, J.W.; Ozols, R.F.; Rose, P.G.; Markman, M.; Armstrong, D.K.; Muggia, F.; McGuire, W.P.; et al. Prognostic factors for stage III epithelial ovarian cancer: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2007, 25, 3621–3627. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Itamochi, H. Neoadjuvant chemotherapy in advanced ovarian cancer: Latest results and place in therapy. Ther. Adv. Med. Oncol. 2014, 6, 293–304. [Google Scholar] [CrossRef]
- Wright, A.A.; Bohlke, K.; Armstrong, D.K.; Bookman, M.A.; Cliby, W.A.; Coleman, R.L.; Dizon, D.S.; Kash, J.J.; Meyer, L.A.; Moore, K.N.; et al. Neoadjuvant Chemotherapy for Newly Diagnosed, Advanced Ovarian Cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 3460–3473. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lee, M.; Kim, H.S.; Chung, H.H.; Song, Y.S. Effect of neoadjuvant chemotherapy on platinum resistance in stage IIIC and IV epithelial ovarian cancer. Medicine 2016, 95, e4797. [Google Scholar] [CrossRef]
- Vergote, I.; Tropé, C.G.; Amant, F.; Kristensen, G.B.; Ehlen, T.; Johnson, N.; Verheijen, R.H.; van der Burg, M.E.; Lacave, A.J.; Panici, P.B.; et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med. 2010, 363, 943–953. [Google Scholar] [CrossRef]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T.; Luesley, D.; Perren, T.; Bannoo, S.; Mascarenhas, M.; et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- Melamed, A.; Fink, G.; Wright, A.A.; Keating, N.L.; Gockley, A.A.; Del Carmen, M.G.; Schorge, J.O.; Rauh-Hain, J.A. Effect of adoption of neoadjuvant chemotherapy for advanced ovarian cancer on all cause mortality: Quasi-experimental study. BMJ 2018, 360, j5463. [Google Scholar] [CrossRef]
- Bristow, R.E.; Chi, D.S. Platinum-based neoadjuvant chemotherapy and interval surgical cytoreduction for advanced ovarian cancer: A meta-analysis. Gynecol. Oncol. 2006, 103, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Sehouli, J.; Savvatis, K.; Braicu, E.I.; Schmidt, S.C.; Lichtenegger, W.; Fotopoulou, C. Primary versus interval debulking surgery in advanced ovarian cancer: Results from a systematic single-center analysis. Int. J. Gynecol. Cancer 2010, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.; Laframboise, S.; Ferguson, S.; Dodge, J.; Bernardini, M.; Murphy, J.; Segev, Y.; Sun, P.; Narod, S.A. The impacts of neoadjuvant chemotherapy and of debulking surgery on survival from advanced ovarian cancer. Gynecol. Oncol. 2014, 134, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.C.; Song, Y.J.; Seo, S.S.; Yoo, C.W.; Kang, S.; Park, S.Y. Residual cancer stem cells after interval cytoreductive surgery following neoadjuvant chemotherapy could result in poor treatment outcomes for ovarian cancer. Oncol. Res. Treat. 2010, 33, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Eno, M.L.; Im, D.D.; Rosenshein, N.B. Chemotherapy time interval and development of platinum and taxane resistance in ovarian, fallopian, and peritoneal carcinomas. Arch. Gynecol. Obstet. 2010, 281, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzales-Martin, A.; Colombo, N.; Sessa, C.; ESMO Guidelines Working Group. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi24–vi32. [Google Scholar] [CrossRef]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Castellarin, M.; Milne, K.; Zeng, T.; Tse, K.; Mayo, M.; Zhao, Y.; Webb, J.R.; Watson, P.H.; Nelson, B.H.; Holt, R.A. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J. Pathol. 2013, 229, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Roncato, R.; Montico, M.; De Mattia, E.; Gagno, S.; Poletto, E.; Scalone, S.; Canzonieri, V.; Giorda, G.; Sorio, R.; et al. New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells 2019, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Matulonis, U. Next-Generation Sequencing: Role in Gynecologic Cancers. J. Natl. Compr. Cancer Netw. 2016, 14, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Cecchin, E.; Canzonieri, V.; Sorio, R.; Giorda, G.; Scalone, S.; De Mattia, E.; Roncato, R.; Gagno, S.; Poletto, E.; et al. Identification of Novel Somatic TP53 Mutations in Patients with High-Grade Serous Ovarian Cancer (HGSOC) Using Next-Generation Sequencing (NGS). Int. J. Mol. Sci. 2018, 19, 1510. [Google Scholar] [CrossRef]
- Suarez-Kelly, L.P.; Akagi, K.; Reeser, J.W.; Samorodnitsky, E.; Reeder, M.; Smith, A.; Roychowdhury, S.; Symer, D.E.; Carson, W.E. Metaplastic breast cancer in a patient with neurofibromatosis type 1 and somatic loss of heterozygosity. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Xie, M.; Wendl, M.C.; Wang, J.; McLellan, M.D.; Leiserson, M.D.; Huang, K.L.; Wyczalkowski, M.A.; Jayasinghe, R.; Banerjee, T.; et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 2015, 6, 10086. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Flaman, J.M.; Frebourg, T.; Moreau, V.; Charbonnier, F.; Martin, C.; Chappuis, P.; Sappino, A.P.; Limacher, I.M.; Bron, L.; Benhattar, J.; et al. A simple p53 functional assay for screening cell lines, blood, and tumors. Proc. Natl. Acad. Sci. USA 1995, 92, 3963–3967. [Google Scholar] [CrossRef]
- Smardova, J.; Liskova, K.; Ravcukova, B.; Malcikova, J.; Hausnerova, J.; Svitakova, M.; Hrabalkova, R.; Zlamalikova, L.; Stano-Kozubik, K.; Blahakova, I.; et al. Complex analysis of the p53 tumor suppressor in lung carcinoma. Oncol. Rep. 2016, 35, 1859–1867. [Google Scholar] [CrossRef]
- Dumont, P.; Leu, J.J.; Della Pietra, A.C., III; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Jeong, B.S.; Hu, W.; Belyi, V.; Rabadan, R.; Levine, A.J. Differential levels of transcription of p53-regulated genes by the arginine/proline polymorphism: p53 with arginine at codon 72 favors apoptosis. FASEB J. 2010, 24, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Zięba, S.; Kowalik, A.; Zalewski, K.; Rusetska, N.; Goryca, K.; Piaścik, A.; Misiek, M.; Bakuła-Zalewska, E.; Kopczyński, J.; Kowalski, K.; et al. Somatic mutation profiling of vulvar cancer: Exploring therapeutic targets. Gynecol. Oncol. 2018, 150, 552–561. [Google Scholar] [CrossRef]
- Kyndi, M.; Alsner, J.; Hansen, L.L.; Sørensen, F.B.; Overgaard, J. LOH rather than genotypes of TP53 codon 72 is associated with disease-free survival in primary breast cancer. Acta Oncol. 2006, 45, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, G.; Biason, P.; Russo, A.; De Mattia, E.; Cecchin, E.; Hattinger, C.M.; Pasello, M.; Alberghini, M.; Ferrari, C.; Scotlandi, K.; et al. Effect of TP53 Arg72Pro and MDM2 SNP309 polymorphisms on the risk of high-grade osteosarcoma development and survival. Clin. Cancer Res. 2009, 15, 3550–3556. [Google Scholar] [CrossRef] [PubMed]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 is a promising molecular target in the diagnosis of cancer. Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef]
- Taube, E.T.; Denkert, C.; Sehouli, J.; Kunze, C.A.; Dietel, M.; Braicu, I.; Letsch, A.; Darb-Esfahani, S. Wilms tumor protein 1 (WT1)—Not only a diagnostic but also a prognostic marker in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2016, 140, 494–502. [Google Scholar] [CrossRef]
- Köbel, M.; Bak, J.; Bertelsen, B.I.; Carpen, O.; Grove, A.; Hansen, E.S.; Levin Jakobsen, A.M.; Lidang, M.; Måsbäck, A.; Tolf, A.; et al. Ovarian carcinoma histotype determination is highly reproducible, and is improved through the use of immunohistochemistry. Histopathology 2014, 64, 1004–1013. [Google Scholar] [CrossRef]
- Oaknin, A.; Guarch, R.; Barretina, P.; Hardisson, D.; González-Martín, A.; Matías-Guiu, X.; Pérez-Fidalgo, A.; Vieites, B.; Romero, I.; Palacios, J. Recommendations for biomarker testing in epithelial ovarian cancer: A National Consensus Statement by the Spanish Society of Pathology and the Spanish Society of Medical Oncology. Clin. Transl. Oncol. 2018, 20, 274–285. [Google Scholar] [CrossRef]
- Netinatsunthorn, W.; Hanprasertpong, J.; Dechsukhum, C.; Leetanaporn, R.; Geater, A. WT1 gene expression as a prognostic marker in advanced serous epithelial ovarian carcinoma: An immunohistochemical study. BMC Cancer 2006, 6, 90. [Google Scholar] [CrossRef]
- Faltas, B.M.; Prandi, D.; Tagawa, S.T.; Molina, A.M.; Nanus, D.M.; Sternberg, C.; Rosenberg, J.; Mosquera, J.M.; Robinson, B.; Elemento, O.; et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet. 2016, 48, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Abbosh, P.; Keliher, D.; Reardon, B.; Miao, D.; Mouw, K.; Weiner-Taylor, A.; Wankowicz, S.; Han, G.; Teo, M.Y.; et al. Mutational patterns in chemotherapy resistant muscle-invasive bladder cancer. Nat. Commun. 2017, 8, 2193. [Google Scholar] [CrossRef]
- Arend, R.C.; Londoño, A.I.; Montgomery, A.M.; Smith, H.J.; Dobbin, Z.C.; Katre, A.A.; Martinez, A.; Yang, E.S.; Alvarez, R.D.; Huh, W.K.; et al. Molecular Response to Neoadjuvant Chemotherapy in High-Grade Serous Ovarian Carcinoma. Mol. Cancer Res. 2018, 16, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, I.; Bosanquet, A.G.; Hermann, S.; Güner, D.; Dörken, B.; Daniel, P.T. Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging chemotherapy. Cell Death Differ. 2003, 10, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Stano-Kozubik, K.; Tichy, B.; Kantorova, B.; Pavlova, S.; Tom, N.; Radova, L.; Smardova, J.; Pardy, F.; Doubek, M.; et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 2015, 29, 877–885. [Google Scholar] [CrossRef]
- Amin, N.A.; Seymour, E.; Saiya-Cork, K.; Parkin, B.; Shedden, K.; Malek, S.N. A Quantitative Analysis of Subclonal and Clonal Gene Mutations before and after Therapy in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2016, 22, 4525–4535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lønning, P.E.; Knappskog, S. Mapping genetic alterations causing chemoresistance in cancer: Identifying the roads by tracking the drivers. Oncogene 2013, 32, 5315–5330. [Google Scholar] [CrossRef] [PubMed]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum- and taxane-based standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef]
- Phillips, K.A.; Nichol, K.; Ozcelik, H.; Knight, J.; Done, S.J.; Goodwin, P.J.; Andrulis, I.L. Frequency of p53 mutations in breast carcinomas from Ashkenazi Jewish carriers of BRCA1 mutations. J. Natl. Cancer Inst. 1999, 91, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.P.; Ahmadian, A.; Pontén, F.; Nistér, M.; Berg, C.; Lundeberg, J.; Uhlén, M.; Pontén, J. Benign clonal keratinocyte patches with p53 mutations show no genetic link to synchronous squamous cell precancer or cancer in human skin. Am. J. Pathol. 1997, 150, 1791–1803. [Google Scholar] [PubMed]
- Lu, M.L.; Wikman, F.; Orntoft, T.F.; Charytonowicz, E.; Rabbani, F.; Zhang, Z.; Dalbagni, G.; Pohar, K.S.; Yu, G.; Cordon-Cardo, C. Impact of alterations affecting the p53 pathway in bladder cancer on clinical outcome, assessed by conventional and array-based methods. Clin. Cancer Res. 2002, 8, 171–179. [Google Scholar] [PubMed]
- Boonstra, J.J.; van der Velden, A.W.; Beerens, E.C.; van Marion, R.; Morita-Fujimura, Y.; Matsui, Y.; Nishihira, T.; Tselepis, C.; Hainaut, P.; Lowe, A.W.; et al. Mistaken identity of widely used esophageal adenocarcinoma cell line TE-7. Cancer Res. 2007, 67, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Galic, V.; Willner, J.; Wollan, M.; Garg, R.; Garcia, R.; Goff, B.A.; Gray, H.J.; Swisher, E.M. Common polymorphisms in TP53 and MDM2 and the relationship to TP53 mutations and clinical outcomes in women with ovarian and peritoneal carcinomas. Genes Chromosom. Cancer 2007, 46, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef]
- Skinner, H.D.; Sandulache, V.C.; Ow, T.J.; Meyn, R.E.; Yordy, J.S.; Beadle, B.M.; Fitzgerald, A.L.; Giri, U.; Ang, K.K.; Myers, J.N. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin. Cancer Res. 2012, 18, 290–300. [Google Scholar] [CrossRef]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef]
- Senturk, S.; Yao, Z.; Camiolo, M.; Stiles, B.; Rathod, T.; Walsh, A.M.; Nemajerova, A.; Lazzara, M.J.; Altorki, N.K.; Krainer, A.; et al. p53Ψ is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc. Natl. Acad. Sci. USA 2014, 111, E3287–E3296. [Google Scholar] [CrossRef]
- Shirole, N.H.; Pal, D.; Kastenhuber, E.R.; Senturk, S.; Boroda, J.; Pisterzi, P.; Miller, M.; Munoz, G.; Anderluh, M.; Ladanyi, M.; et al. TP53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions. Elife 2017, 6, e25532. [Google Scholar] [CrossRef]
- Smeby, J.; Sveen, A.; Eilertsen, I.A.; Danielsen, S.A.; Hoff, A.M.; Eide, P.W.; Johannessen, B.; Hektoen, M.; Skotheim, R.I.; Guren, M.G.; et al. Transcriptional and functional consequences of TP53 splice mutations in colorectal cancer. Oncogenesis 2019, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, N.; Friedlander, P.; Chen, X.; Prives, C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 2002, 21, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, J.K.; Feng, S.; Dalgarno, D.C.; Brauer, A.W.; Schreiber, S.L. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 1994, 76, 933–945. [Google Scholar] [CrossRef]
- Walker, K.K.; Levine, A.J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Natl. Acad. Sci. USA 1996, 93, 15335–15340. [Google Scholar] [CrossRef] [Green Version]
- Bonafé, M.; Ceccarelli, C.; Farabegoli, F.; Santini, D.; Taffurelli, M.; Barbi, C.; Marzi, E.; Trapassi, C.; Storci, G.; Olivieri, F.; et al. Retention of the p53 codon 72 arginine allele is associated with a reduction of disease-free and overall survival in arginine/proline heterozygous breast cancer patients. Clin. Cancer Res. 2003, 9, 4860–4864. [Google Scholar]
- Xu, Y.; Yao, L.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Lin, B.; Lu, Y.; Xie, Y. p53 Codon 72 polymorphism predicts the pathologic response to neoadjuvant chemotherapy in patients with breast cancer. Clin. Cancer Res. 2005, 11, 7328–7333. [Google Scholar] [CrossRef]
- Toyama, T.; Zhang, Z.; Nishio, M.; Hamaguchi, M.; Kondo, N.; Iwase, H.; Iwata, H.; Takahashi, S.; Yamashita, H.; Fujii, Y. Association of TP53 codon 72 polymorphism and the outcome of adjuvant therapy in breast cancer patients. Breast Cancer Res. 2007, 9, R34. [Google Scholar] [CrossRef]
- Carter, H.; Marty, R.; Hofree, M.; Gross, A.M.; Jensen, J.; Fisch, K.M.; Wu, X.; DeBoever, C.; Van Nostrand, E.L.; Song, Y.; et al. Interaction Landscape of Inherited Polymorphisms with Somatic Events in Cancer. Cancer Discov. 2017, 7, 410–423. [Google Scholar] [CrossRef]
- Galvan, A.; Ioannidis, J.P.; Dragani, T.A. Beyond genome-wide association studies: Genetic heterogeneity and individual predisposition to cancer. Trends Genet. 2010, 26, 132–141. [Google Scholar] [CrossRef]
- Li, Q.; Seo, J.H.; Stranger, B.; McKenna, A.; Pe’er, I.; Laframboise, T.; Brown, M.; Tyekucheva, S.; Freedman, M.L. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013, 152, 633–641. [Google Scholar] [CrossRef]
- Kanchi, K.L.; Johnson, K.J.; Lu, C.; McLellan, M.D.; Leiserson, M.D.; Wendl, M.C.; Zhang, Q.; Koboldt, D.C.; Xie, M.; Kandoth, C.; et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014, 5, 3156. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C. DNA copy number variation and loss of heterozygosity in relation to recurrence of and survival from head and neck squamous cell carcinoma: A review. Head Neck 2008, 30, 1361–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weren, R.D.; Mensenkamp, A.R.; Simons, M.; Eijkelenboom, A.; Sie, A.S.; Ouchene, H.; van Asseldonk, M.; Gomez-Garcia, E.B.; Blok, M.J.; de Hullu, J.A.; et al. Novel BRCA1 and BRCA2 Tumor Test as Basis for Treatment Decisions and Referral for Genetic Counselling of Patients with Ovarian Carcinomas. Hum. Mutat. 2017, 38, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Khiabanian, H.; Hirshfield, K.M.; Goldfinger, M.; Bird, S.; Stein, M.; Aisner, J.; Toppmeyer, D.; Wong, S.; Chan, N.; Dhar, K.; et al. Inference of Germline Mutational Status and Evaluation of Loss of Heterozygosity in High-Depth, Tumor-Only Sequencing Data. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Garg, K.; Levine, D.A.; Olvera, N.; Dao, F.; Bisogna, M.; Secord, A.A.; Berchuck, A.; Cerami, E.; Schultz, N.; Soslow, R.A. BRCA1 immunohistochemistry in a molecularly characterized cohort of ovarian high-grade serous carcinomas. Am. J. Surg. Pathol. 2013, 37, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Chen, J.; Weiss, W.A. Alternative splicing in cancer: Implications for biology and therapy. Oncogene 2015, 34, 1–14. [Google Scholar] [CrossRef]
- de Jonge, M.M.; Ruano, D.; van Eijk, R.; van der Stoep, N.; Nielsen, M.; Wijnen, J.T.; Ter Haar, N.T.; Baalbergen, A.; Bos, M.E.M.M.; Kagie, M.J.; et al. Validation and Implementation of BRCA1/2 Variant Screening in Ovarian Tumor Tissue. J. Mol. Diagn. 2018, 20, 600–611. [Google Scholar] [CrossRef]
- Dubbink, H.J.; Atmodimedjo, P.N.; van Marion, R.; Krol, N.M.G.; Riegman, P.H.J.; Kros, J.M.; van den Bent, M.J.; Dinjens, W.N.M. Diagnostic Detection of Allelic Losses and Imbalances by Next-Generation Sequencing: 1p/19q Co-Deletion Analysis of Gliomas. J. Mol. Diagn. 2016, 18, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Faltas, B.M. Treatment resistance in urothelial carcinoma: An evolutionary perspective. Nat. Rev. Clin. Oncol. 2018, 15, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Zenz, T.; Kröber, A.; Scherer, K.; Häbe, S.; Bühler, A.; Benner, A.; Denzel, T.; Winkler, D.; Edelmann, J.; Schwänen, C.; et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: Results from a detailed genetic characterization with long-term follow-up. Blood 2008, 112, 3322–3329. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Smardova, J.; Rocnova, L.; Tichy, B.; Kuglik, P.; Vranova, V.; Cejkova, S.; Svitakova, M.; Skuhrova Francova, H.; Brychtova, Y.; et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: Selection, impact on survival, and response to DNA damage. Blood 2009, 114, 5307–5314. [Google Scholar] [CrossRef] [PubMed]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Tothill, R.W.; McArthur, G.A. Navigating the challenge of tumor heterogeneity in cancer therapy. Cancer Discov. 2014, 4, 146–148. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood | Tumor | Tumor | TP53 Gene | Genomic | cDNA | Ref_ | Alt | Variant | AA | Predicted |
|---|---|---|---|---|---|---|---|---|---|---|
| (Germline) | Pre-NACT | Post-NACT | Region | Coordinate * | Change | seq | _seq | Type | Change | Protein Product |
| No | Yes | Yes | IVS4 | 17:7,579,311 | c.375+1G>A | G | A | SD | p.Gly59Valfs*23 | Truncated |
| Yes | Yes | Yes | Exon 4 | 17:7,579,472 | c.215C>G | C | G | Missense | p.Pro72Arg | Full-lenght |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garziera, M.; Cecchin, E.; Giorda, G.; Sorio, R.; Scalone, S.; De Mattia, E.; Roncato, R.; Gagno, S.; Poletto, E.; Romanato, L.; et al. Clonal Evolution of TP53 c.375+1G>A Mutation in Pre- and Post- Neo-Adjuvant Chemotherapy (NACT) Tumor Samples in High-Grade Serous Ovarian Cancer (HGSOC). Cells 2019, 8, 1186. https://doi.org/10.3390/cells8101186
Garziera M, Cecchin E, Giorda G, Sorio R, Scalone S, De Mattia E, Roncato R, Gagno S, Poletto E, Romanato L, et al. Clonal Evolution of TP53 c.375+1G>A Mutation in Pre- and Post- Neo-Adjuvant Chemotherapy (NACT) Tumor Samples in High-Grade Serous Ovarian Cancer (HGSOC). Cells. 2019; 8(10):1186. https://doi.org/10.3390/cells8101186
Chicago/Turabian StyleGarziera, Marica, Erika Cecchin, Giorgio Giorda, Roberto Sorio, Simona Scalone, Elena De Mattia, Rossana Roncato, Sara Gagno, Elena Poletto, Loredana Romanato, and et al. 2019. "Clonal Evolution of TP53 c.375+1G>A Mutation in Pre- and Post- Neo-Adjuvant Chemotherapy (NACT) Tumor Samples in High-Grade Serous Ovarian Cancer (HGSOC)" Cells 8, no. 10: 1186. https://doi.org/10.3390/cells8101186
APA StyleGarziera, M., Cecchin, E., Giorda, G., Sorio, R., Scalone, S., De Mattia, E., Roncato, R., Gagno, S., Poletto, E., Romanato, L., Ecca, F., Canzonieri, V., & Toffoli, G. (2019). Clonal Evolution of TP53 c.375+1G>A Mutation in Pre- and Post- Neo-Adjuvant Chemotherapy (NACT) Tumor Samples in High-Grade Serous Ovarian Cancer (HGSOC). Cells, 8(10), 1186. https://doi.org/10.3390/cells8101186