Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis


{kind=link}
{kind=link}
Abstract
:1. Introduction
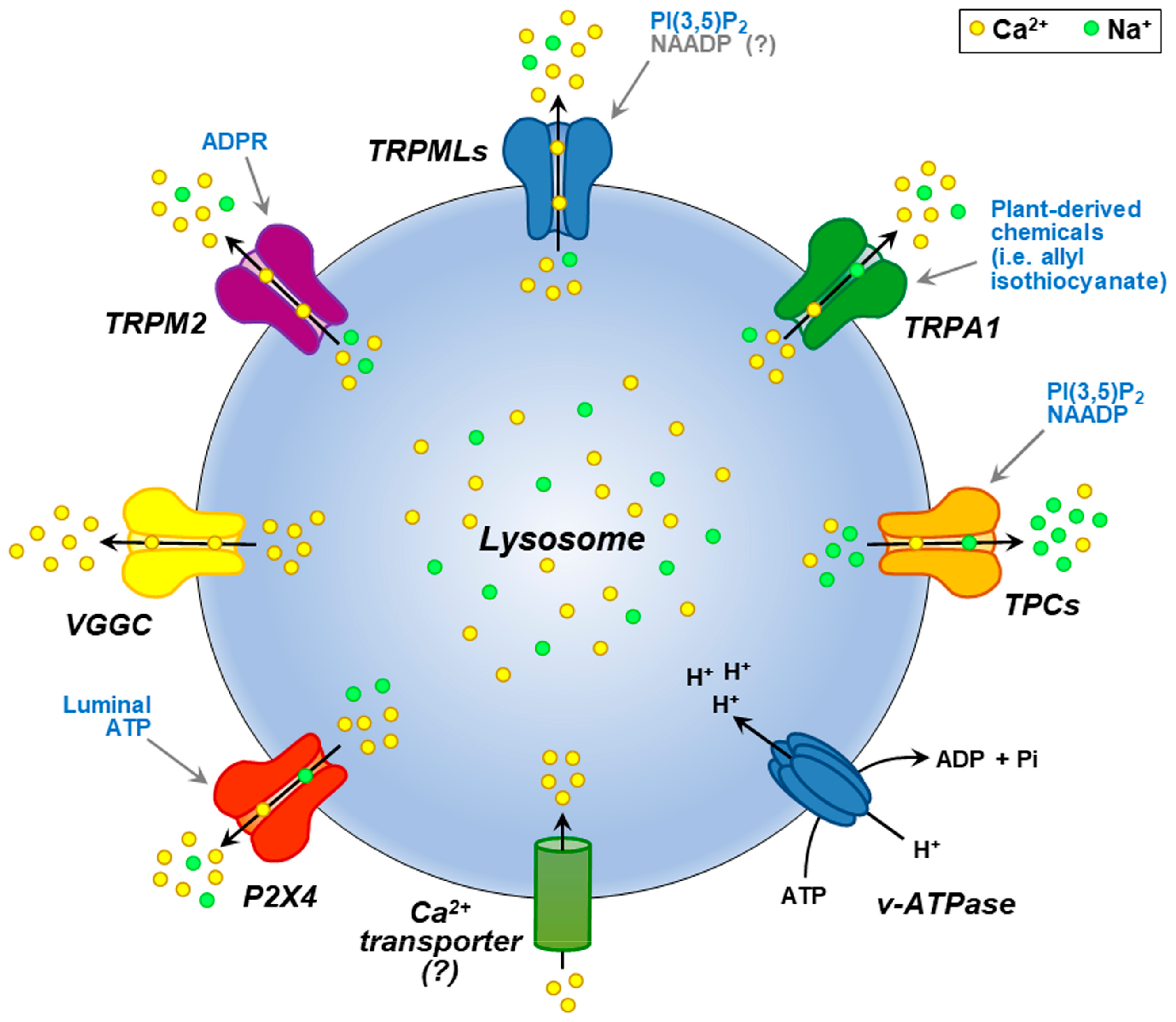
2. Lysosomal Ca2+ within the Global Ca2+ Homeostasis: Interplay and Function
3. Lysosomal Ca2+ Signaling and Autophagy
4. Lysosomal Ca2+ Dysfunction in Neurodegeneration
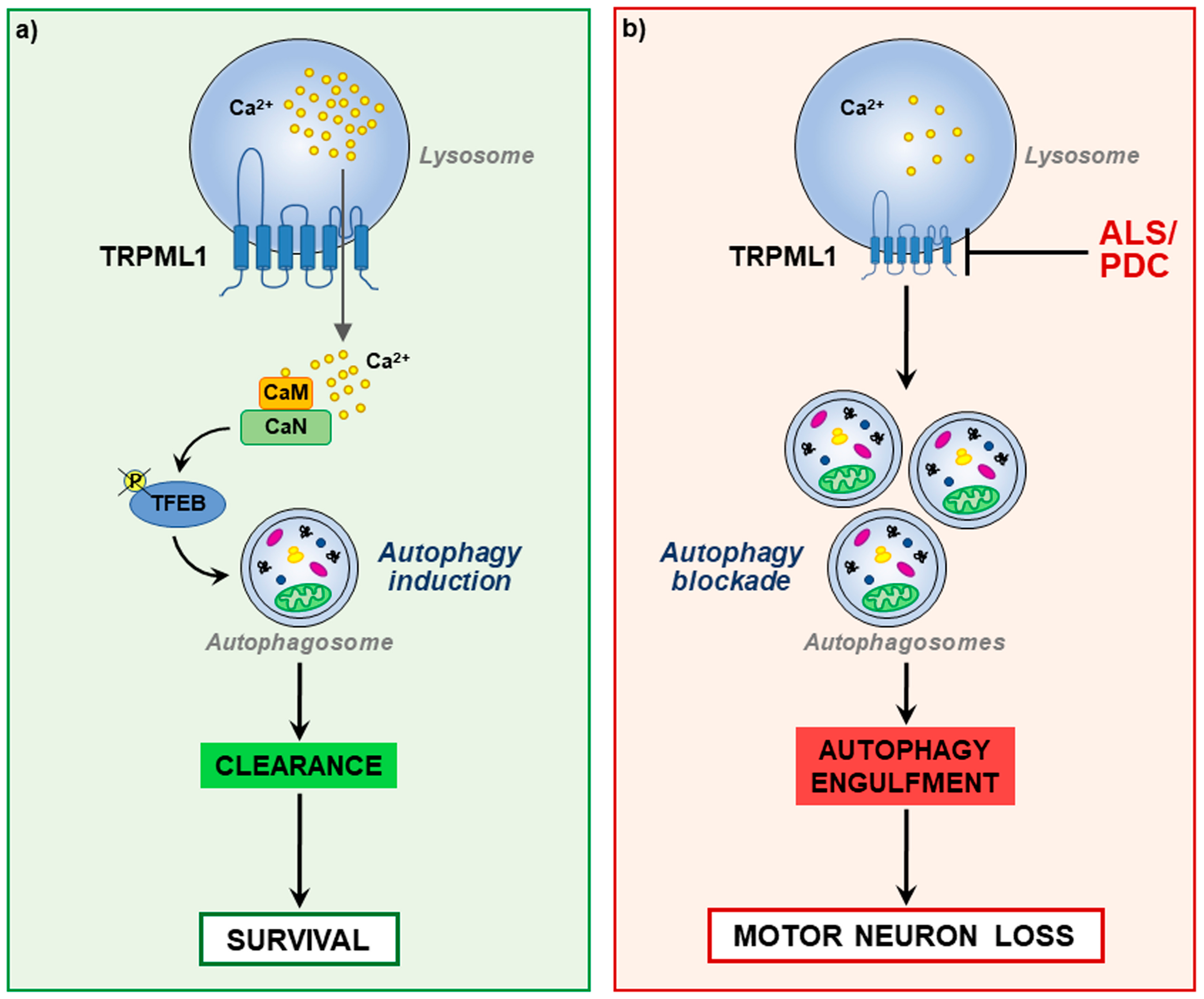
5. Lysosomal Ca2+ Dysfunction and Autophagy Defects in ALS
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Beleza-Meireles, A.; Al-Chalabi, A. Genetic studies of amyotrophic lateral sclerosis: Controversies and perspectives. Amyotroph. Lateral Scler. 2009, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Amyotrophic lateral sclerosis and glutamate-too much of a good thing? N. Engl. J. Med. 1992, 326, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrì, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun. 2017, 483, 1187–1193. [Google Scholar] [CrossRef]
- Li, A.; Ségui, J.; Heinemann, S.H.; Hoshi, T. Oxidation regulates cloned neuronal voltage-dependent Ca2+ channels expressed in Xenopus oocytes. J. Neurosci. 1998, 18, 6740–6747. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Williams, T.L.; Day, N.C.; Ince, P.G.; Kamboj, R.K.; Shaw, P.J. Calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors: A molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 200–207. [Google Scholar] [CrossRef]
- Leal, S.S.; Cardoso, I.; Valentine, J.S.; Gomes, C.M. Calcium ions promote superoxide dismutase 1 (SOD1) aggregation into non-fibrillar amyloid: A link to toxic effects of calcium overload in amyotrophic lateral sclerosis (ALS)? J. Biol. Chem. 2013, 288, 25219–25228. [Google Scholar] [CrossRef]
- Kiselyov, K.; Muallem, S. ROS and intracellular ion channels. Cell Calcium 2016, 60, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Petrozziello, T.; Secondo, A.; Tedeschi, V.; Esposito, A.; Sisalli, M.; Scorziello, A.; Di Renzo, G.; Annunziato, L. ApoSOD1 lacking dismutase activity neuroprotects motor neurons exposed to beta-methylamino-L-alanine through the Ca2+/Akt/ERK1/2 prosurvival pathway. Cell Death Differ. 2017, 24, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, V.; Petrozziello, T.; Sisalli, M.J.; Boscia, F.; Canzoniero, L.M.T.; Secondo, A. The activation of Mucolipin TRP channel 1 (TRPML1) protects motor neurons from L-BMAA neurotoxicity by promoting autophagic clearance. Sci. Rep. 2019, 9, 10743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, J.K. Calcium–How and why? J. Biosci. 2001, 26, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Godinho, L.F.; Costello, J.L.; Islinger, M. The different facets of organelle interplay-an overview of organelle interactions. Front. Cell Dev. Biol. 2015, 3, 56. [Google Scholar] [CrossRef]
- Han, Y.; Li, M.; Qiu, F.; Zhang, M.; Zhang, Y.H. Cell-permeable organic fluorescent probes for live-cell long-term super-resolution imaging reveal lysosome-mitochondrion interactions. Nat. Commun. 2017, 8, 1307. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef]
- Capel, R.A.; Bolton, E.L.; Lin, W.K.; Aston, D.; Wang, Y.; Liu, W.; Wang, X.; Burton, R.A.; Bloor-Young, D.; Shade, K.T.; et al. Two-pore Channels (TPC2s) and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) at Lysosomal-Sarcoplasmic Reticular Junctions Contribute to Acute and Chronic β-Adrenoceptor Signaling in the Heart. J. Biol. Chem. 2015, 290, 30087–30098. [Google Scholar] [CrossRef]
- Kinnear, N.P.; Boittin, F.X.; Thomas, J.M.; Galione, A.; Evans, A.M. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J. Biol. Chem. 2004, 279, 54319–54326. [Google Scholar] [CrossRef] [PubMed]
- Penny, C.J.; Kilpatrick, B.S.; Han, J.M.; Sneyd, J.; Patel, S. A computational model of lysosome-ER Ca2+ microdomains. J. Cell Sci. 2014, 127, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Aston, D.; Capel, R.A.; Ford, K.L.; Christian, H.C.; Mirams, G.R.; Rog-Zielinska, E.A.; Kohl, P.; Galione, A.; Burton, R.A.; Terrar, D.A. High resolution structural evidence suggests the Sarcoplasmic Reticulum forms microdomains with Acidic Stores (lysosomes) in the heart. Sci. Rep. 2017, 7, 40620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [PubMed]
- Bygrave, F.L.; Benedetti, A. What is the concentration of calcium ions in the endoplasmic reticulum? Cell Calcium 1996, 19, 547–551. [Google Scholar] [CrossRef]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Magalhaes, J.; Beavan, M.S.; McNeill, A.; Gegg, M.E.; Cleeter, M.W.; Bloor-Young, D.; Churchill, G.C.; Duchen, M.R.; Schapira, A.H.; et al. Endoplasmic reticulum and lysosomal Ca²⁺ stores are remodelled in GBA1-linked Parkinson disease patient fibroblasts. Cell Calcium 2016, 59, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef]
- Osmanovic, A.; Rangnau, I.; Kosfeld, A.; Abdulla, S.; Janssen, C.; Auber, B.; Raab, P.; Preller, M.; Petri, S.; Weber, R.G. FIG4 variants in central European patients with amyotrophic lateral sclerosis: A whole-exome and targeted sequencing study. Eur. J. Hum. Genet. 2017, 25, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Bargal, R.; Avidan, N.; Ben-Asher, E.; Olender, Z.; Zeigler, M.; Frumkin, A.; Raas-Rothschild, A.; Glusman, G.; Lancet, D.; Bach, G. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 2000, 26, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPlante, J.M.; Falardeau, J.; Sun, M.; Kanazirska, M.; Brown, E.M.; Slaugenhaupt, S.A.; Vassilev, P.M. Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett. 2002, 532, 183–187. [Google Scholar] [CrossRef]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ahuja, M.; Kim, M.S.; Brailoiu, G.C.; Jha, A.; Zeng, M.; Baydyuk, M.; Wu, L.G.; Wassif, C.A.; Porter, F.D.; et al. Fusion of lysosomes with secretory organelles leads to uncontrolled exocytosis in the lysosomal storage disease mucolipidosis type IV. EMBO Rep. 2016, 17, 266–278. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, X.; Gao, Q.; Ali Samie, M.; Azar, M.; Tsang, W.L.; Dong, L.; Sahoo, N.; Li, X.; Zhuo, Y.; et al. The intracellular Ca²⁺ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 2014, 20, 1187–1192. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, E.; Rahman, T.; Churamani, D.; Prole, D.L.; Brailoiu, G.C.; Hooper, R.; Taylor, C.W.; Patel, S. An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J. Biol. Chem. 2010, 285, 38511–38516. [Google Scholar] [CrossRef]
- Zong, X.; Schieder, M.; Cuny, H.; Fenske, S.; Gruner, C.; Rötzer, K.; Griesbeck, O.; Harz, H.; Biel, M.; Wahl-Schott, C. The two-pore channel TPCN2 mediates NAADP-dependent Ca(2+)-release from lysosomal stores. Pflugers Arch. 2009, 458, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Waller-Evans, H.; Peterneva, K.; Platt, F.M. Endolysosomal calcium regulation and disease. Biochem. Soc. Trans. 2010, 38, 1458–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penny, C.J.; Patel, S. Poring over two-pore channel pore mutants. Messenger 2015, 4, 46–52. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef]
- Guo, J.; Zeng, W.; Jiang, Y. Tuning the ion selectivity of two-pore channels. Proc. Natl. Acad. Sci. USA 2017, 114, 1009–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife 2016, 5, e15887. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Michalak, M.; Agellon, L.B. Endoplasmic Reticulum Malfunction in the Nervous System. Front. Neurosci. 2017, 11, 220. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Pavez, M.; Thompson, A.C.; Arnott, H.J.; Mitchell, C.B.; D’Atri, I.; Don, E.K.; Chilton, J.K.; Scott, E.K.; Lin, J.Y.; Young, K.M.; et al. STIM1 Is Required for Remodeling of the Endoplasmic Reticulum and Microtubule Cytoskeleton in Steering Growth Cones. J. Neurosci. 2019, 39, 5095–5114. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Adachi, Y.; Yamada, T.; Suzuki, T.L.; Otomo, T.; McBride, H.M.; Yoshimori, T.; Iijima, M.; Sesaki, H. A brain-enriched Drp1 isoform associates with lysosomes, late endosomes, and the plasma membrane. J. Biol. Chem. 2018, 293, 11809–11822. [Google Scholar] [CrossRef] [Green Version]
- Cousin, M.A.; Robinson, P.J. Ca(2+) influx inhibits dynamin and arrests synaptic vesicle endocytosis at the active zone. J. Neurosci. 2000, 20, 949–957. [Google Scholar] [CrossRef]
- Zou, J.; Hu, B.; Arpag, S.; Yan, Q.; Hamilton, A.; Zeng, Y.S.; Vanoye, C.G.; Li, J. Reactivation of Lysosomal Ca2+ Efflux Rescues Abnormal Lysosomal Storage in FIG4-Deficient Cells. J. Neurosci. 2015, 35, 6801–6812. [Google Scholar] [CrossRef]
- Yoon, Y.; Pitts, K.R.; McNiven, M.A. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol. Biol. Cell 2001, 12, 2894–2905. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Zhou, J.; Liu, W.; Duan, X.; Gala, U.; Sandoval, H.; Jaiswal, M.; Tong, C. Dynamin Regulates Autophagy by Modulating Lysosomal Function. J. Genet. Genomics 2016, 43, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Durchfort, N.; Verhoef, S.; Vaughn, M.B.; Shrestha, R.; Adam, D.; Kaplan, J.; Ward, D.M. The enlarged lysosomes in beige j cells result from decreased lysosome fission and not increased lysosome fusion. Traffic 2012, 13, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Yates, E.; Futter, C.E.; Patel, S. An Endosomal NAADP-Sensitive Two-Pore Ca2+ Channel Regulates ER-Endosome Membrane Contact Sites to Control Growth Factor Signaling. Cell Rep. 2017, 18, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Chaperone-mediated autophagy: Selectivity pays off. Trends Endocrinol. Metab. 2010, 21, 142–150. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Integration of clearance mechanisms: The proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2010, 2, a006734. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb. Perspect. Biol. 2012, 4, a008839. [Google Scholar] [CrossRef]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Ballabio, A. Lysosomal calcium regulates autophagy. Autophagy 2015, 11, 970–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergarajauregui, S.; Connelly, P.S.; Daniels, M.P.; Puertollano, R. Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet. 2008, 17, 2723–2737. [Google Scholar] [CrossRef]
- Curcio-Morelli, C.; Charles, F.A.; Micsenyi, M.C.; Cao, Y.; Venugopal, B.; Browning, M.F.; Dobrenis, K.; Cotman, S.L.; Walkley, S.U.; Slaugenhaupt, S.A. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 2010, 40, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Long, A.A.; Elsaesser, R.; Nikolaeva, D.; Broadie, K.; Montell, C. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell 2008, 135, 838–851. [Google Scholar] [CrossRef]
- Sun, T.; Wang, X.; Lu, Q.; Ren, H.; Zhang, H. CUP-5, the C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy 2011, 7, 1308–1315. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.T.; Manzoni, M.; Monti, E.; Pizzo, M.T.; Ballabio, A.; Borsani, G. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am. J. Hum. Genet. 2000, 67, 1110–1120. [Google Scholar] [CrossRef]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, N.; Lang, M.J.; Weisman, L.S. Phosphatidylinositol 3,5-bisphosphate: Regulation of cellular events in space and time. Biochem. Soc. Trans. 2016, 44, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Patel, N.; Xu, H.; Lee, M.; Tominaga-Yamanaka, K.; Nath, A.; Geiger, J.; Gorospe, M.; Mattson, M.P.; Haughey, N.J. Activation of TRPML1 clears intraneuronal Aβ in preclinical models of HIV infection. J. Neurosci. 2014, 34, 11485–11503. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca(2+) homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205. [Google Scholar] [CrossRef]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Nagarkar Jaiswal, S.; di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; Duraine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Kaski, D.; Hanna, M.G. Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat. Rev. Neurol. 2012, 8, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Orellana, F.; Godoy, P.A.; Silva-Grecchi, T.; Barra, K.M.; Fuentealba, J. Modulation of the neuronal network activity by P2X receptors and their involvement in neurological disorders. Pharmacol. Res. 2015, 101, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington’s disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Tsuji, S.; Takahashi, H. Involvement of lysosomes in the pathogenesis of CAG repeat diseases. Ann. Neurol. 2002, 52, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Kim, H.N.; Hwang, J.J.; Kim, Y.H.; Park, S.E. Lysosomal dysfunction in proteinopathic neurodegenerative disorders: Possible therapeutic roles of cAMP and zinc. Mol. Brain 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.; Xu, H.; Zeng, Z.; Zhu, H. TRPML1 Participates in the Progression of Alzheimer’s Disease by Regulating the PPARγ/AMPK/Mtor Signalling Pathway. Cell. Physiol. Biochem. 2017, 43, 2446–2456. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef]
- Tsunemi, T.; Perez-Rosello, T.; Ishiguro, Y.; Yoroisaka, A.; Jeon, S.; Hamada, K.; Rammonhan, M.; Wong, Y.C.; Xie, Z.; Akamatsu, W.; et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca2+ Channel Agonists Protects Human Dopaminergic Neurons from α-Synuclein Toxicity. J. Neurosci. 2019, 39, 5760–5772. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol. Dis. 2019, 122, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Cipolat Mis, M.S.; Brajkovic, S.; Frattini, E.; Di Fonzo, A.; Corti, S. Autophagy in motor neuron disease: Key pathogenetic mechanisms and therapeutic targets. Mol. Cell. Neurosci. 2016, 72, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 2015, 125, 2548. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Medinas, D.B.; Rozas, P.; Martínez Traub, F.; Woehlbier, U.; Brown, R.H.; Bosco, D.A.; Hetz, C. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 8209–8214. [Google Scholar] [CrossRef] [Green Version]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibiger, C.; Deisel, J.; Aufschnaiter, A.; Ambros, S.; Tereshchenko, M.; Verheijen, B.M.; Büttner, S.; Braun, R.J. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum. Mol. Genet. 2018, 27, 1593–1607. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, N.; Nagai, M.; Ohta, Y.; Miyazaki, K.; Kurata, T.; Morimoto, M.; Murakami, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T.; et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007, 1167, 112–117. [Google Scholar] [CrossRef]
- Zhang, Y.; Burberry, A.; Wang, J.Y.; Sandoe, J.; Ghosh, S.; Udeshi, N.D.; Svinkina, T.; Mordes, D.A.; Mok, J.; Charlton, M.; et al. The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev. 2018, 32, 929–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugolino, J.; Ji, Y.J.; Conchina, K.; Chu, J.; Nirujogi, R.S.; Pandey, A.; Brady, N.R.; Hamacher-Brady, A.; Wang, J. Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLoS Genet. 2016, 12, e1006443. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Varghese, A.M.; Vijaylakshmi, K.; Sumitha, R.; Prasanna, V.K.; Shruthi, S.; Chandrasekhar Sagar, B.K.; Datta, K.K.; Gowda, H.; Nalini, A.; et al. Cerebrospinal Fluid from Sporadic Amyotrophic Lateral Sclerosis Patients Induces Mitochondrial and Lysosomal Dysfunction. Neurochem. Res. 2016, 41, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, J.T.; Sechi, A.; Dreser, A.; Katona, I.; Wiemuth, D.; Vervoorts, J.; Dohmen, M.; Chandrasekar, A.; Prause, J.; Brauers, E.; et al. Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis. 2014, 5, e1290. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Petrozziello, T.; Tedeschi, V.; Esposito, A.; Secondo, A. Pharmacology of Amyotrophic Lateral Sclerosis: Old Strategies and New Perspectives. In Drug Repositioning: Approaches and Applications for Neurotherapeutics, 1st ed.; Dudley, J., Berliocchi, L., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 261–274. ISBN 9781315373669. [Google Scholar]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tedeschi, V.; Petrozziello, T.; Secondo, A. Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1216. https://doi.org/10.3390/cells8101216
Tedeschi V, Petrozziello T, Secondo A. Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis. Cells. 2019; 8(10):1216. https://doi.org/10.3390/cells8101216
Chicago/Turabian StyleTedeschi, Valentina, Tiziana Petrozziello, and Agnese Secondo. 2019. "Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis" Cells 8, no. 10: 1216. https://doi.org/10.3390/cells8101216
APA StyleTedeschi, V., Petrozziello, T., & Secondo, A. (2019). Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis. Cells, 8(10), 1216. https://doi.org/10.3390/cells8101216