Emerging Approaches to Investigate the Influence of Transition Metals in the Proteinopathies

 , , , , , , ,
, , , , , , ,  and
and
Abstract
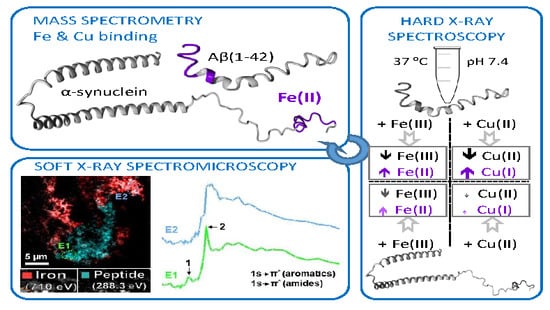
:
1. Introduction
2. Materials and Methods
2.1. Materials for In-Vitro Experiments
2.1.1. For XANES Experiments
2.1.2. For MS Experiments
2.1.3. For STXM Experiments
2.2. Human Tissue
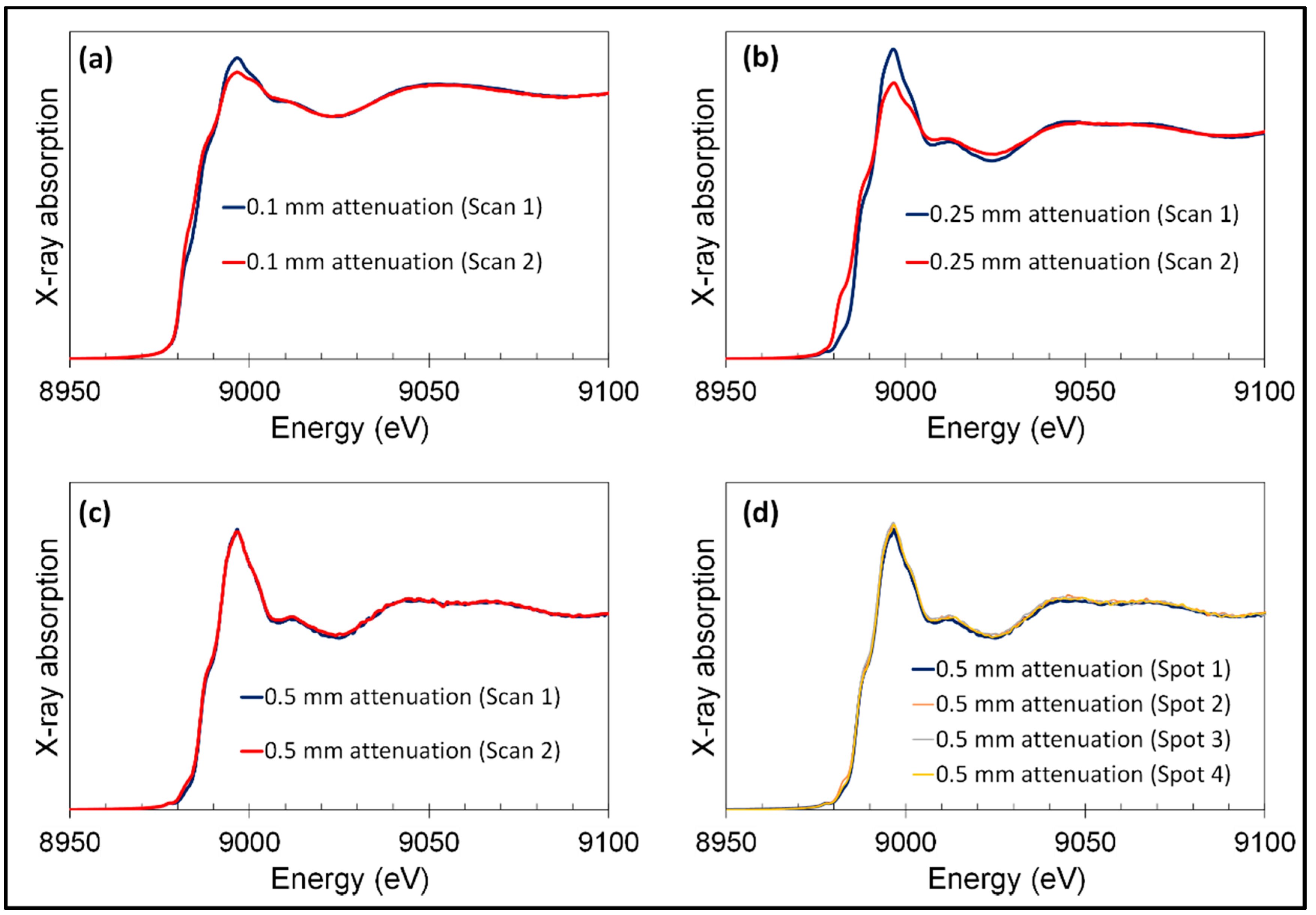
2.3. Hard X-ray Spectromicroscopy
2.3.1. For μXRF
2.3.2. For XANES
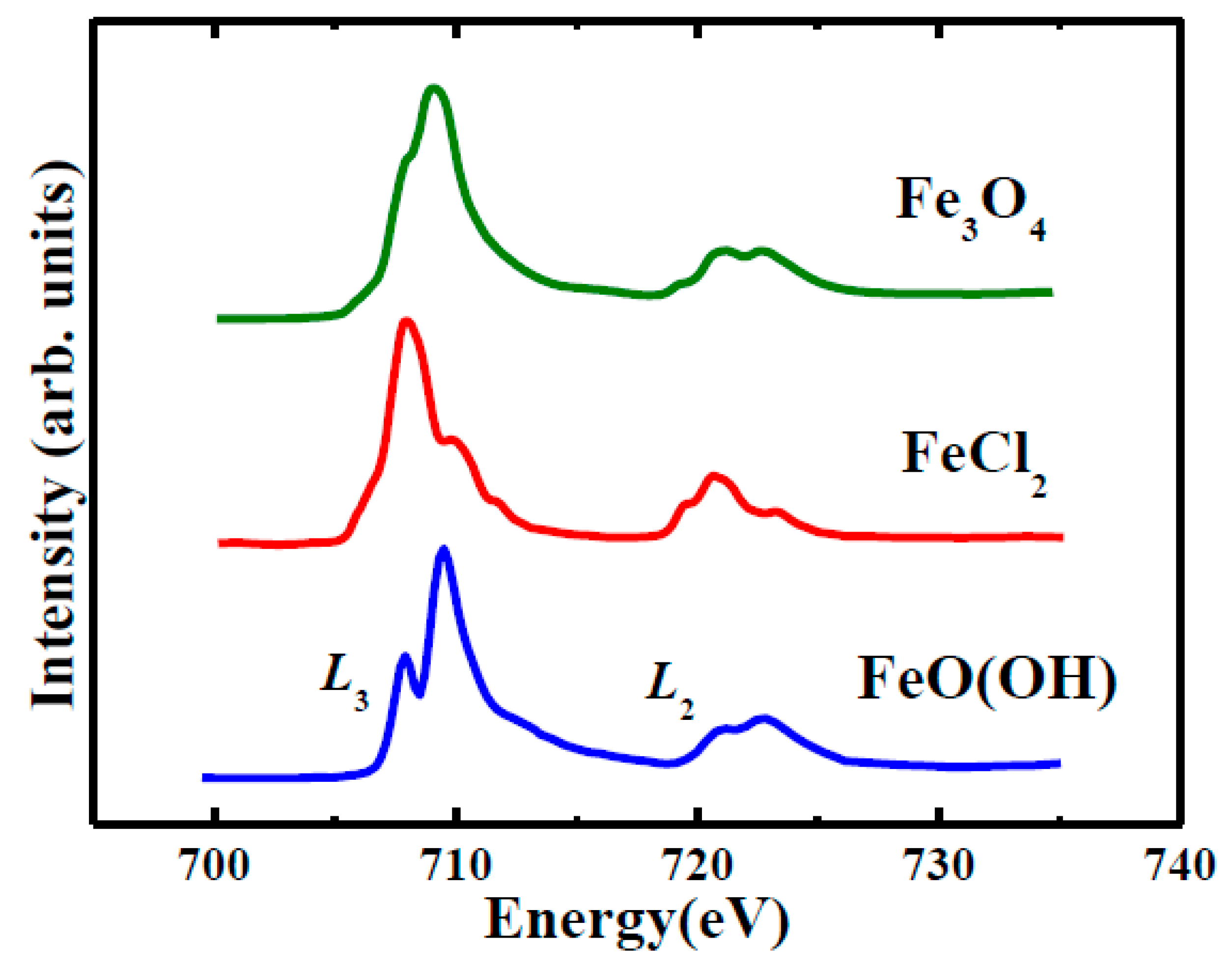
2.4. Soft X-ray Spectromicroscopy
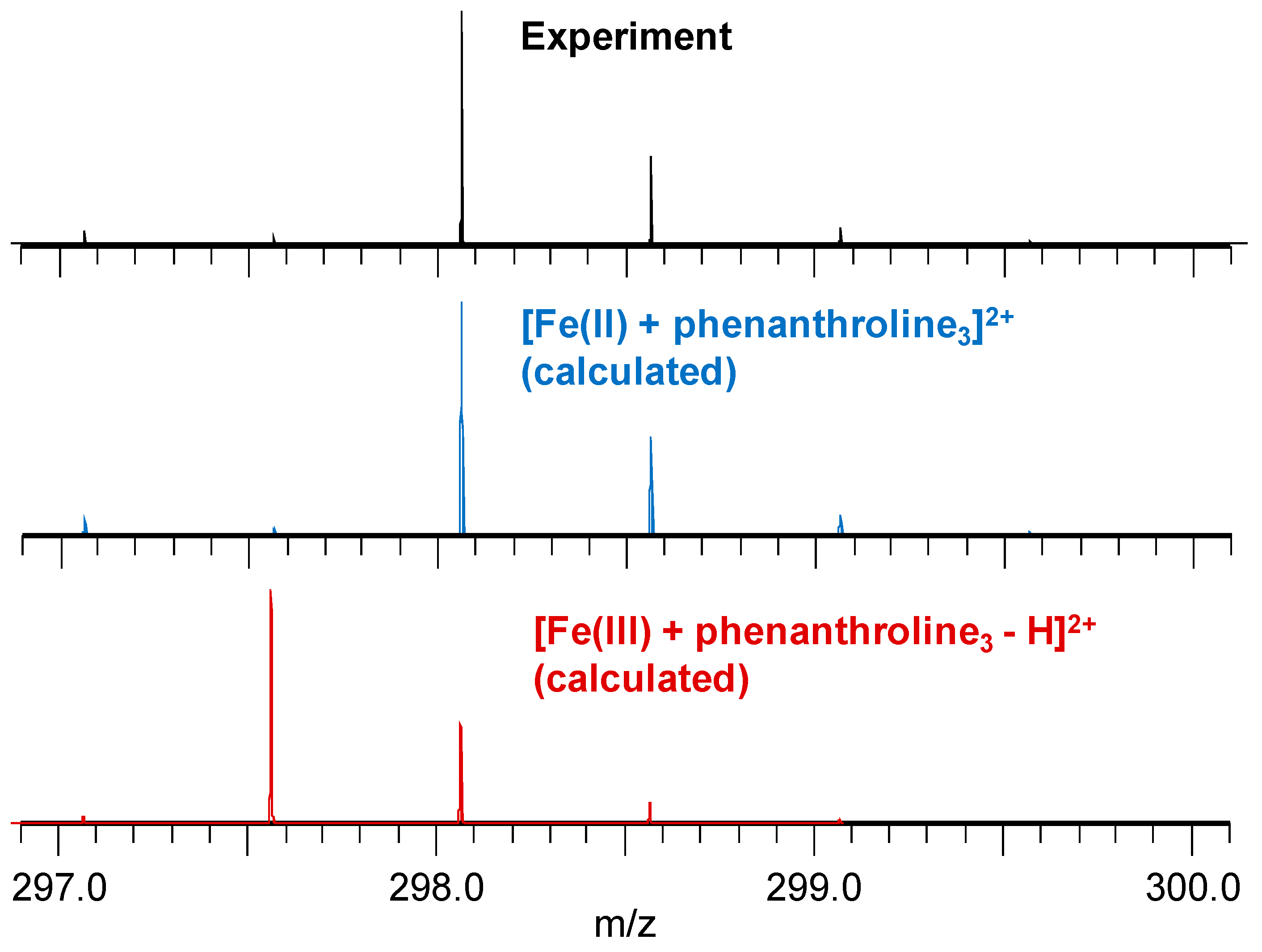
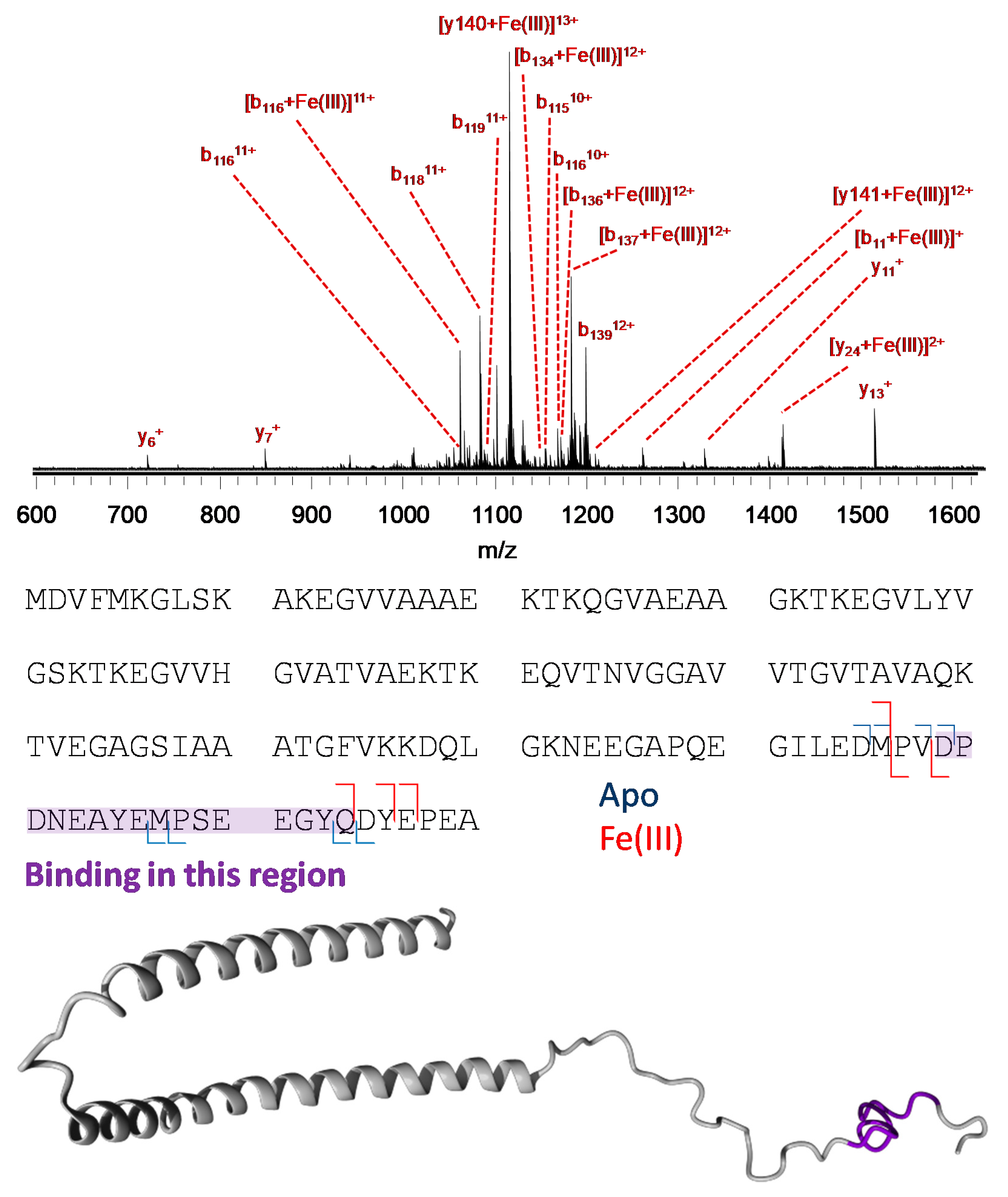
2.5. Mass Spectrometry
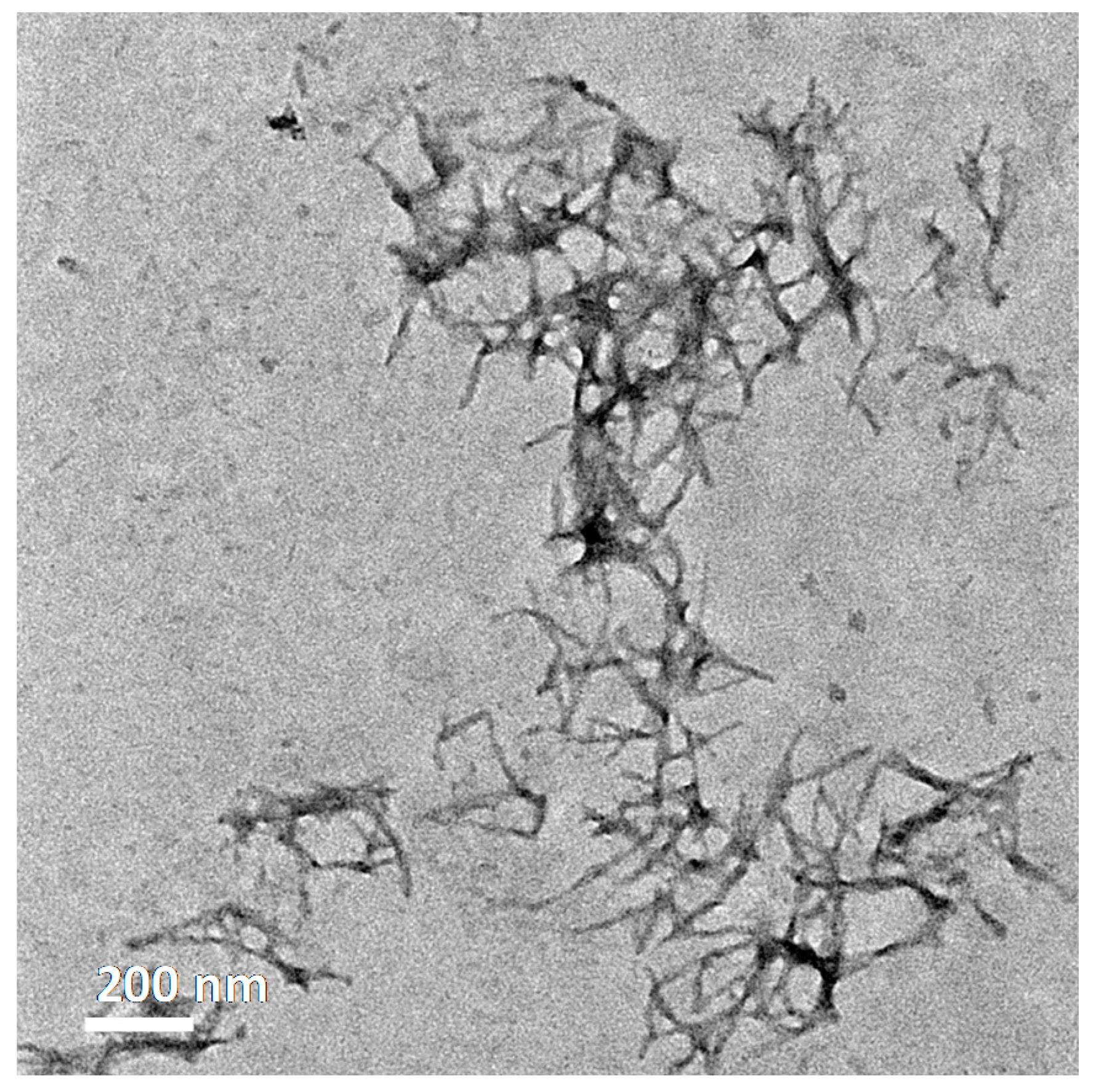
2.6. Transmission Electron Microscopy
3. Results
3.1. Hard X-Ray Analysis
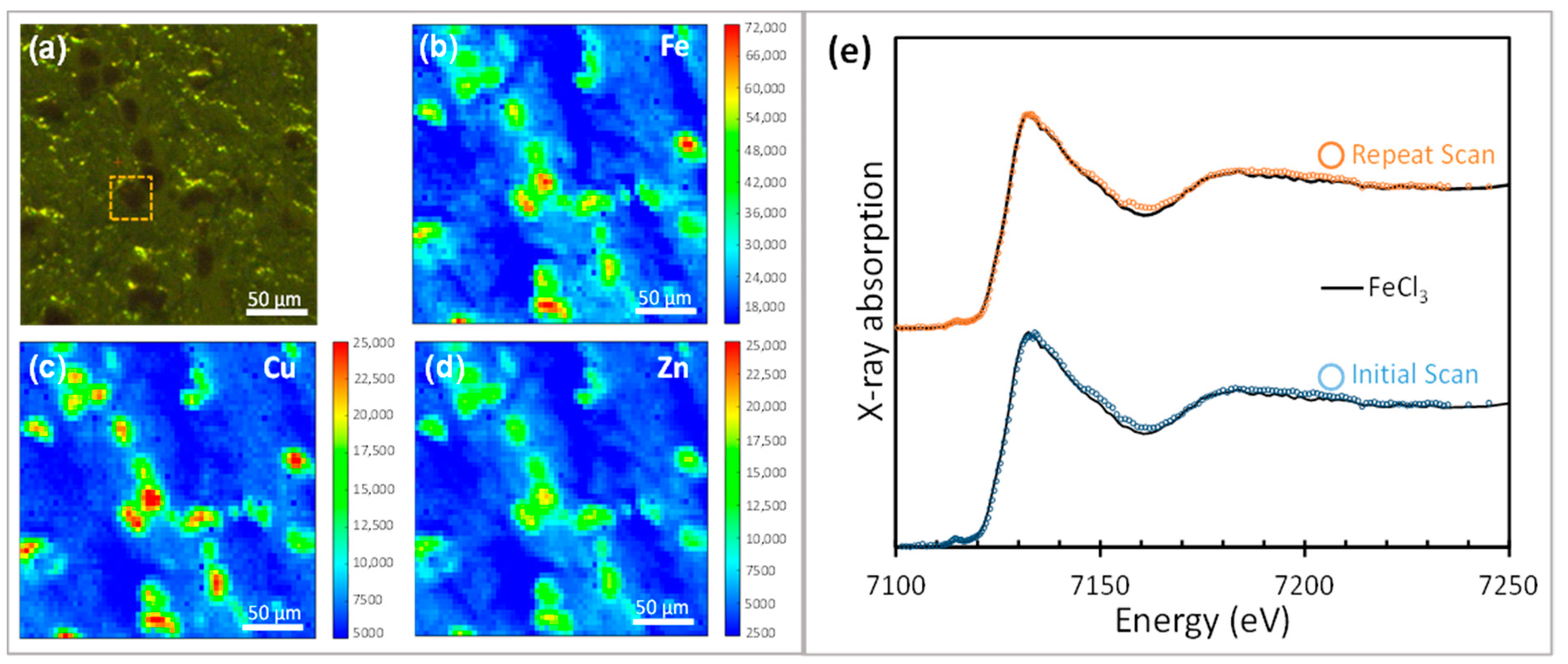
3.1.1. XANES of Iron in Human Brain Tissue
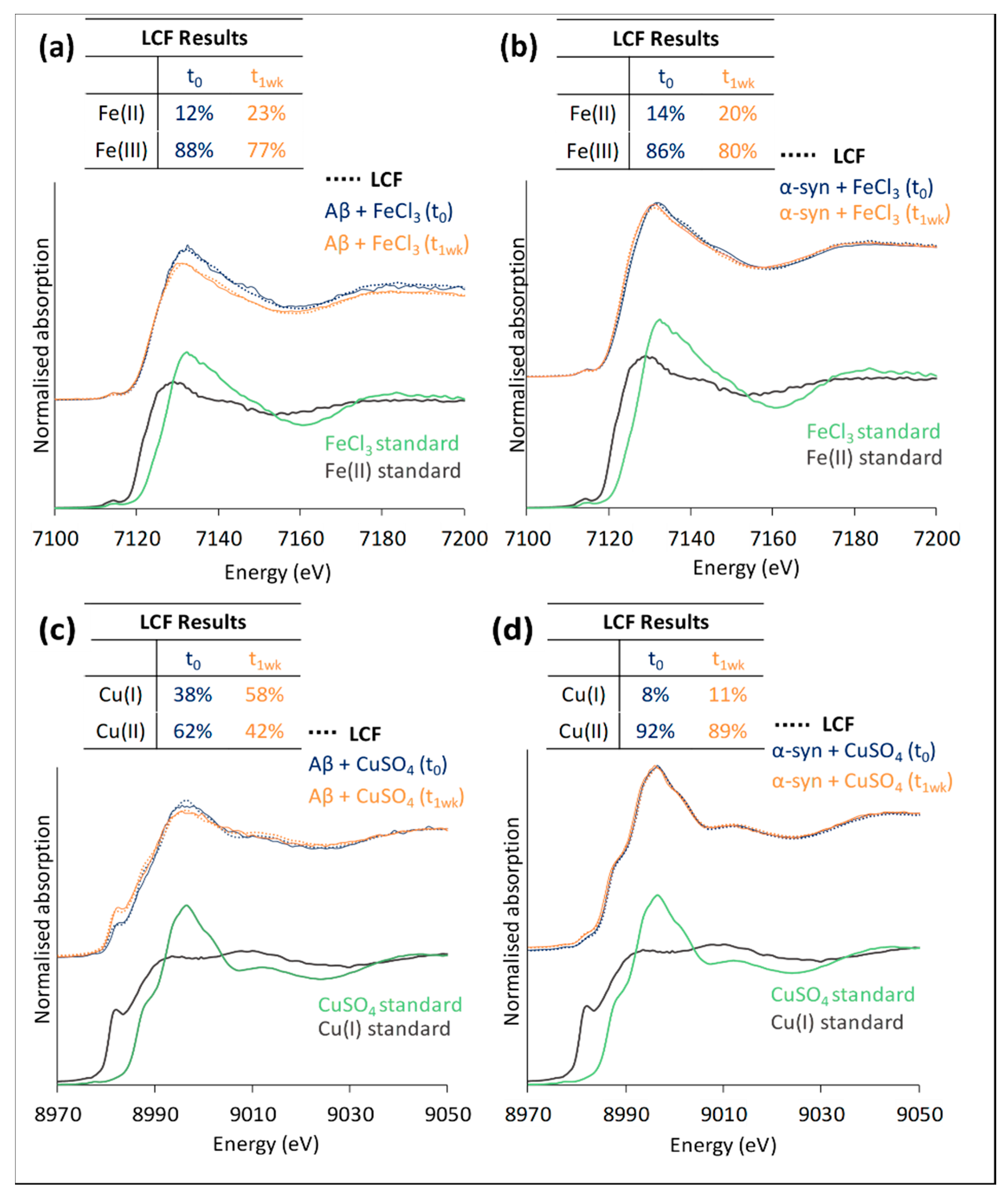
3.1.2. XANES of Iron and Copper in In-Vitro Systems
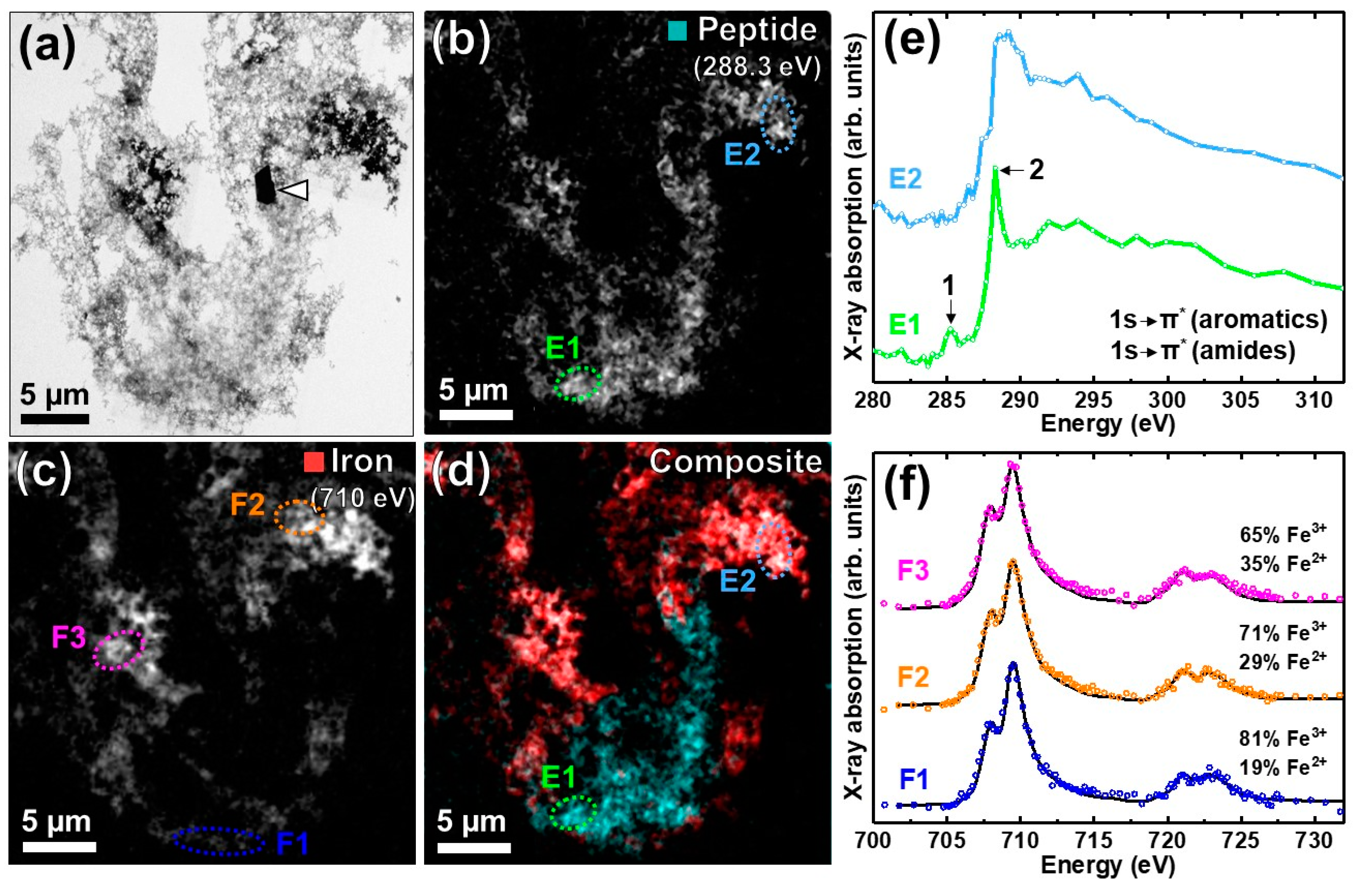
3.2. Nanoscale Chemical Speciation of Aβ/Iron Aggregate Structures Formed In-Vitro
3.2.1. Amyloid β(1–42) and Iron(III) In-Vitro
3.2.2. STXM Spectromicroscopy of Aβ(1–42) and Iron(III) In Vitro
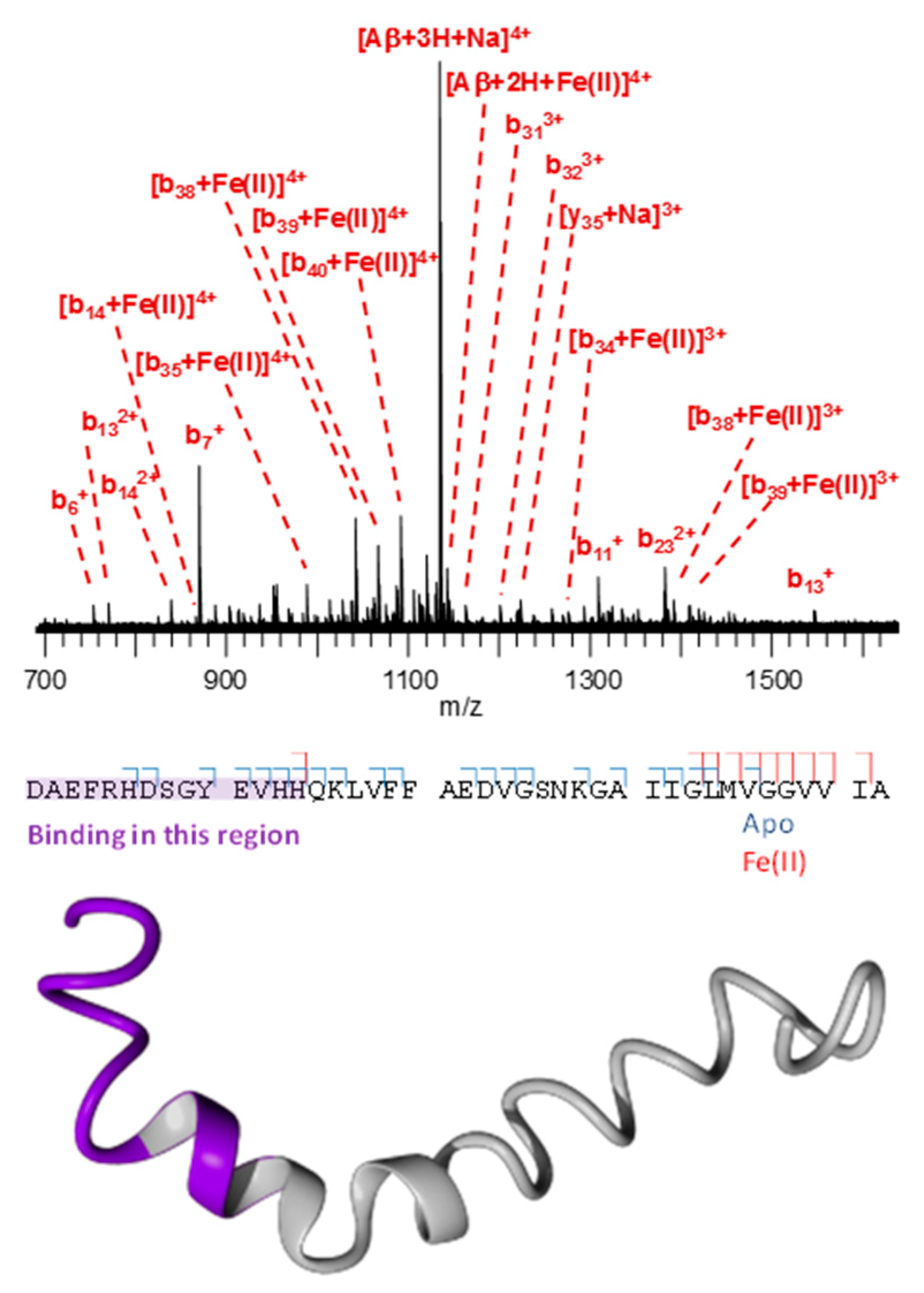
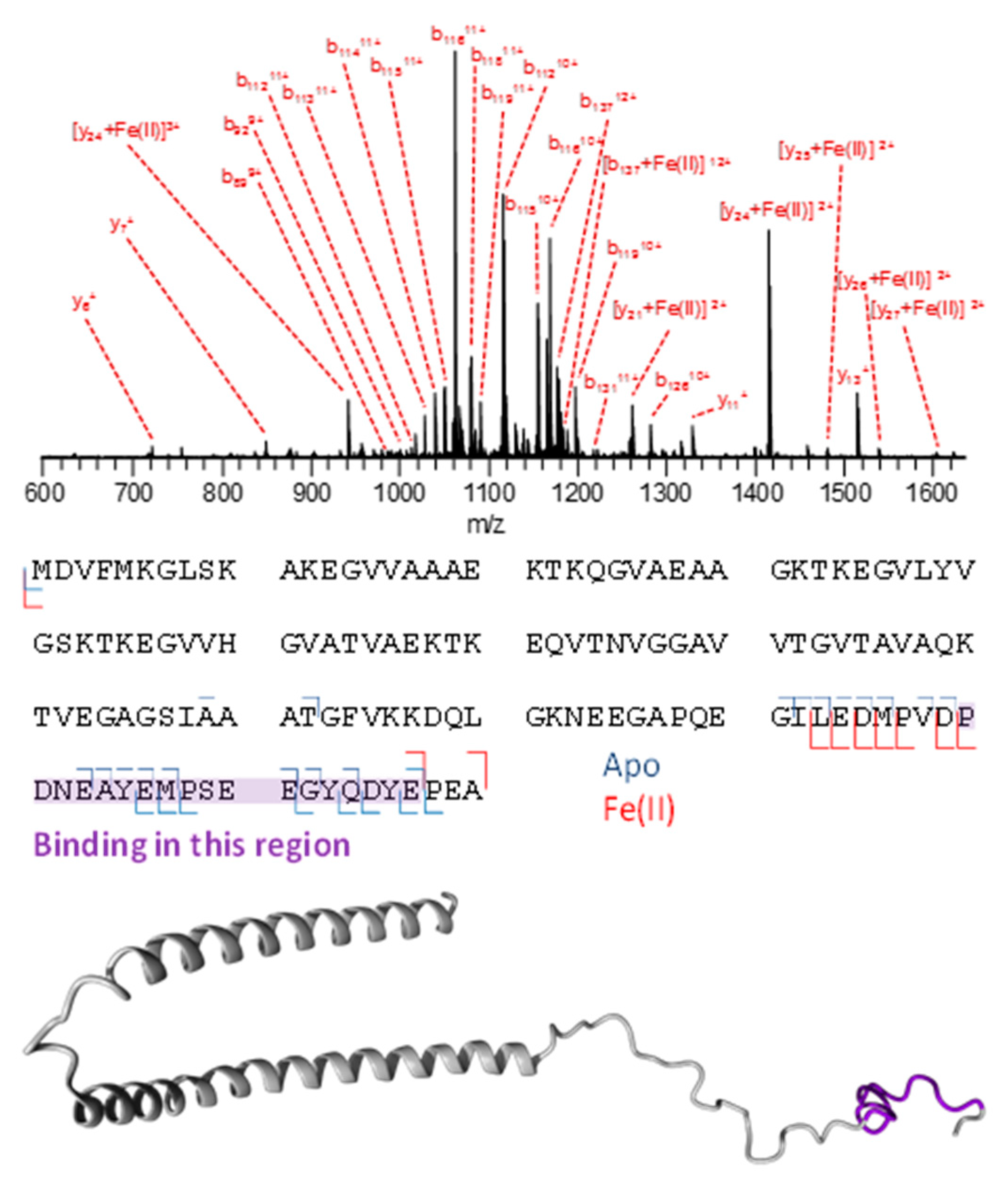
3.3. Mass Spectrometry Allowed Determination of the Specific Binding Regions in [α-syn + Cu], [α-syn + Fe], [Aβ + Cu], and [Aβ + Fe] Complexes
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Mass Spectrometry




Appendix B. Soft X-Ray Spectromicroscopy


Appendix C. Hard X-Ray Beam Optimization to Avoid Beam-Induced Photo-Reduction

Appendix D. Mass Spectrometry Peak Assignment Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | Exact m/z | Observed m/z | Error (ppm) |
|---|---|---|---|
| [y6]+ | 723.2832 | 723.2835 | 0.3 |
| [y7]+ | 851.3418 | 851.342 | 0.3 |
| [b116]11+ | 1062.6557 | 1062.6562 | 0.4 |
| [b116+Fe(III)]11+ | 1067.4659 | 1067.4663 | 0.4 |
| [b118]11+ | 1080.4849 | 1080.4854 | 0.4 |
| [b119]11+ | 1090.9419 | 1090.9433 | 1.2 |
| [y140+Fe(III)]13+ | 1116.7096 | 1116.71 | 0.3 |
| [b134+Fe(III)]12+ | 1149.4952 | 1149.4948 | −0.4 |
| [b115]10+ | 1155.7165 | 1155.7171 | 0.5 |
| [b116]10+ | 1168.8206 | 1168.8209 | 0.3 |
| [b136+Fe(III)]12+ | 1172.6694 | 1172.6705 | 0.9 |
| [b137+Fe(III)]12+ | 1183.4229 | 1183.4232 | 0.2 |
| [y22+Fe(III)]2+ | 1315.9455 | 1315.9458 | 0.2 |
| [y24+Fe(III)]2+ | 1414.0061 | 1414.0067 | 0.4 |
| [y13]+ | 1513.5966 | 1513.5967 | 0.1 |
| [y14]+ | 1644.6371 | 1644.6362 | −0.5 |
| Average | 0.32 | ||
| Standard Dev | 0.41 |
| Ion | Exact m/z | Observed m/z | Error (ppm) |
|---|---|---|---|
| [y3]+ | 316.1504 | 316.1504 | 0.3 |
| [y4]+ | 445.193 | 445.1929 | −0.2 |
| [y6]+ | 723.2832 | 723.2834 | 0.3 |
| [y7]+ | 851.3418 | 851.3426 | 1.0 |
| [y24+Fe(II)]3+ | 943.3424 | 943.343 | 0.6 |
| [b89]9+ | 981.2003 | 981.2009 | 0.6 |
| [b118]12+ | 990.5285 | 990.5304 | 2.0 |
| [b92]9+ | 1008.2138 | 1008.215 | 1.2 |
| [b112]11+ | 1018.2745 | 1018.2755 | 1.0 |
| [b113]11+ | 1028.5548 | 1028.5555 | 0.7 |
| [b114]11+ | 1040.286 | 1040.2868 | 0.8 |
| [b115]11+ | 1050.743 | 1050.7432 | 0.2 |
| [b116]11+ | 1062.6557 | 1062.6561 | 0.3 |
| [y9]+ | 1071.4266 | 1071.4268 | 0.1 |
| [b127]12+ | 1079.2265 | 1079.2263 | −0.2 |
| [b118]11+ | 1080.4849 | 1080.4851 | 0.1 |
| [b119]11+ | 1090.9419 | 1090.9421 | 0.1 |
| [y28+Fe(II)]3+ | 1106.0738 | 1106.0726 | −1.0 |
| [b111]10+ | 1108.6928 | 1108.6937 | 0.8 |
| [y140]13+ | 1112.6395 | 1112.6405 | 0.9 |
| [b140+Fe(II)]13+ | 1115.4017 | 1115.4025 | 0.7 |
| [y140+Fe(II)]13+ | 1116.7872 | 1116.7873 | 0.1 |
| [b112]10+ | 1120.0012 | 1120.0012 | 0.0 |
| [b113]10+ | 1131.3096 | 1131.3099 | 0.3 |
| [b123]11+ | 1132.3206 | 1132.3218 | 1.1 |
| [b124]11+ | 1138.7785 | 1138.7784 | −0.1 |
| [b114]10+ | 1144.2139 | 1144.2139 | 0.0 |
| [b125]11+ | 1153.6025 | 1153.6022 | −0.2 |
| [b115]10+ | 1155.7165 | 1155.7166 | 0.0 |
| [b126]11+ | 1165.3336 | 1165.3337 | 0.1 |
| [b116]10+ | 1168.8206 | 1168.8206 | 0.0 |
| [b137]12+ | 1179.0136 | 1179.0136 | 0.0 |
| [b137+Fe(II)]12+ | 1183.5069 | 1183.5072 | 0.2 |
| [b118]10+ | 1188.4327 | 1188.4326 | −0.1 |
| [b119]10+ | 1199.9354 | 1199.9355 | 0.0 |
| [b131]11+ | 1217.4436 | 1217.4437 | 0.0 |
| [b132]11+ | 1222.6274 | 1222.6259 | −1.2 |
| [b134]11+ | 1249.093 | 1249.0937 | 0.6 |
| [b124]10+ | 1252.5556 | 1252.5544 | −1.0 |
| [y21+Fe(II)]2+ | 1258.9359 | 1258.9364 | 0.4 |
| [b126]10+ | 1281.7662 | 1281.7663 | 0.0 |
| [b127]10+ | 1294.8703 | 1294.8713 | 0.8 |
| [b116]9+ | 1298.5776 | 1298.578 | 0.3 |
| [y22+Fe(II)]2+ | 1316.4494 | 1316.4494 | −0.1 |
| [y24+Fe(II)]2+ | 1414.51 | 1414.5111 | 0.8 |
| [y25+Fe(II)]2+ | 1480.0303 | 1480.0317 | 1.0 |
| [y13]+ | 1513.5966 | 1513.5984 | 1.2 |
| [y26+Fe(II)]2+ | 1537.5437 | 1537.5462 | 1.6 |
| [y27+Fe(II)]2+ | 1602.065 | 1602.0663 | 0.8 |
| [y14]+ | 1644.6371 | 1644.6396 | 1.5 |
| [y15]+ | 1773.6796 | 1773.679 | −0.3 |
| Average | 0.36 | ||
| Standard Dev | 0.63 |
| Ion | Exact m/z | Observed m/z | Error (ppm) |
|---|---|---|---|
| [b6]+ | 756.3424 | 756.3424 | 0.0 |
| [b13]2+ | 772.3317 | 772.3321 | 0.5 |
| [b14]2+ | 840.8612 | 840.8609 | −0.3 |
| [b14+Fe(II)]2+ | 867.8208 | 867.8203 | −0.6 |
| [b7]+ | 871.3694 | 871.3693 | −0.1 |
| [b15]2+ | 904.8904 | 904.8903 | −0.1 |
| [b33]4+ | 914.4486 | 914.4484 | −0.3 |
| [b23]3+ | 920.0930 | 920.0917 | −1.5 |
| [b33+Fe(II)]4+ | 927.9284 | 927.9271 | −1.4 |
| [b24]3+ | 953.1158 | 953.1152 | −0.7 |
| [b34+Fe(II)]4+ | 956.1994 | 956.1987 | −0.8 |
| [b16]2+ | 968.9379 | 968.9357 | −2.3 |
| [b25]3+ | 972.1230 | 972.1219 | −1.1 |
| [b35+Fe(II)]4+ | 988.9595 | 988.9590 | −0.5 |
| [b36+Fe(II)]4+ | 1013.7267 | 1013.7269 | 0.3 |
| [b37+Fe(II)]4+ | 1027.9820 | 1027.9809 | −1.1 |
| [b38+Fe(II)]4+ | 1042.2374 | 1042.2368 | −0.6 |
| [b39+Fe(II)]4+ | 1067.0045 | 1067.0040 | −0.5 |
| [b18]2+ | 1075.0142 | 1075.0129 | −1.1 |
| [b28]3+ | 1081.8463 | 1081.8454 | −0.8 |
| [b40+Fe(II)]4+ | 1091.7716 | 1091.7710 | −0.5 |
| [b41+Fe(II)]4+ | 1120.0426 | 1120.0429 | 0.3 |
| [b30]3+ | 1124.5325 | 1124.5312 | −1.2 |
| [Aβ+3H+Na]4+ | 1134.3202 | 1134.3200 | −0.2 |
| [b42+Fe(II)]4+ | 1137.8019 | 1137.8008 | −1.0 |
| [Aβ+2H+Fe(II)]4+ | 1142.3045 | 1142.3048 | 0.2 |
| [b19]2+ | 1148.5484 | 1148.5461 | −2.0 |
| [b31]3+ | 1162.2272 | 1162.2275 | 0.3 |
| [b10]+ | 1178.4862 | 1178.4860 | −0.2 |
| [b32]3+ | 1199.9219 | 1199.9220 | 0.1 |
| [y35+Na]3+ | 1221.9705 | 1221.9705 | 0.0 |
| [b33+Fe(II)]3+ | 1236.9021 | 1236.9009 | −1.0 |
| [b34]3+ | 1256.6237 | 1256.6254 | 1.3 |
| [b34+Fe(II)]3+ | 1274.5968 | 1274.5957 | −0.9 |
| [b11]+ | 1307.5288 | 1307.5297 | 0.7 |
| [b35+Fe(II)]3+ | 1318.2770 | 1318.2779 | 0.7 |
| [b22]2+ | 1322.1224 | 1322.1225 | 0.0 |
| [b36]3+ | 1333.3267 | 1333.3287 | 1.5 |
| [b23]2+ | 1379.6359 | 1379.6386 | 2.0 |
| [b38+Fe(II)]3+ | 1389.3141 | 1389.3165 | 1.7 |
| [b12]+ | 1406.5972 | 1406.5977 | 0.3 |
| [b39+Fe(II)]3+ | 1422.3369 | 1422.3377 | 0.6 |
| [b24]2+ | 1429.1701 | 1429.1705 | 0.2 |
| [b40+Fe(II)]3+ | 1455.3597 | 1455.3625 | 1.9 |
| [b13]+ | 1543.6561 | 1543.6578 | 1.1 |
| Average | −0.16 | ||
| Standard Dev | 0.97 |
| Ion | Exact m/z | Observed m/z | Error (ppm) |
|---|---|---|---|
| [c6]+ | 769.3736 | 769.3736 | 0.0 |
| [c9]+ | 1026.5111 | 1026.5095 | 1.6 |
| [c17+Cu(II)]2+ | 935.4487 | 935.4486 | 0.1 |
| [c22+Cu(I)]2+ | 1186.0824 | 1186.0817 | 0.5 |
| [c28+Cu(I)]2+ | 1492.2439 | 1492.2419 | 1.4 |
| [c31+Cu(I)]2+ | 1591.7918 | 1591.7892 | 1.6 |
| [c23+Cu(II)]3+ | 833.4197 | 833.4196 | 0.2 |
| [c28+Cu(II)]3+ | 994.8291 | 994.8276 | 1.5 |
| [c31+Cu(II)]3+ | 1061.1943 | 1061.1922 | 2.0 |
| [c33+Cu(I)]3+ | 1137.9112 | 1137.9096 | 1.4 |
| [c35+Cu(I)]3+ | 1223.6237 | 1223.6213 | 1.9 |
| [c38+Cu(I)]3+ | 1313.3483 | 1313.3462 | 1.6 |
| [c39+Cu(I)]3+ | 1367.7028 | 1367.7026 | 0.1 |
| [c46+Cu(I)]3+ | 1610.8368 | 1610.8379 | −0.7 |
| [c38+Cu(I)]4+ | 985.2631 | 985.2613 | 1.8 |
| [c39+Cu(I)]4+ | 1026.0289 | 1026.0269 | 1.9 |
| [c50+Cu(II)]5+ | 1045.1469 | 1045.1460 | 0.9 |
| [c57+Cu(II)]5+ | 1170.6114 | 1170.6118 | −0.3 |
| [c61+Cu(I)]5+ | 1268.0690 | 1268.0696 | −0.4 |
| [c68+Cu(II)]7+ | 999.5258 | 999.5248 | 1.1 |
| [c75+Cu(II)]7+ | 1089.1486 | 1089.1479 | 0.6 |
| [c75]7+ | 1080.4466 | 1080.4454 | 1.1 |
| [c95+Cu(II)]9+ | 1050.5581 | 1050.5583 | −0.2 |
| [c96+Cu(II)]9+ | 1064.7909 | 1064.7905 | 0.4 |
| [c139+Cu(I)]11+ | 1313.8364 | 1313.8377 | −1.0 |
| [z4+H]+ | 429.1748 | 429.1749 | −0.3 |
| [z46+H]3+ | 1723.0915 | 1723.0913 | 0.1 |
| [z45+H]3+ | 1690.0687 | 1690.0696 | −0.5 |
| [z57+H]4+ | 1518.4317 | 1518.4329 | −0.8 |
| [z65+H]5+ | 1380.2378 | 1380.2380 | −0.2 |
| [z102+H]8+ | 1323.1481 | 1323.1489 | −0.6 |
| [z101+H]8+ | 1302.7652 | 1302.7661 | −0.8 |
| [z113]9+ | 1296.5308 | 1296.5318 | −0.8 |
| [z112+H]9+ | 1282.3046 | 1282.3062 | −1.2 |
| [z102+H]9+ | 1176.2435 | 1176.2431 | 0.4 |
| [z140+Cu(I)]10+ | 1450.6188 | 1450.6190 | −0.1 |
| [z140+Cu(I)]11+ | 1318.8360 | 1318.8374 | −1.1 |
| [z140+Cu(I)]12+ | 1209.0169 | 1209.0177 | −0.7 |
| Average | 0.33 | ||
| Standard Dev | 0.98 |
References
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. The amyloid cascade hypothesis in Alzheimer’s disease: It’s time to change our mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized trial of verubecestat for mild-to-moderate alzheimer’s disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Chang, C.J. Searching for harmony in transition-metal signaling. Nat. Chem. Biol. 2015, 11, 744. [Google Scholar] [CrossRef] [PubMed]
- Collingwood, J.F.; Adams, F. Chemical imaging analysis of the brain with X-ray methods. Spectrochim. Acta Part B At. Spectrosc. 2017, 130, 101–118. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease: How far have we come in the clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem 2016, 139, 179–197. [Google Scholar] [CrossRef]
- Nunez, M.T.; Chana-Cuevas, P. New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 2018, 11, 109. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, J.; Collingwood, J.F.; Tjendana-Tjhin, V.; Brooks, J.; Lermyte, F.; Plascencia-Villa, G.; Hands-Portman, I.; Dobson, J.; Perry, G.; Telling, N.D. Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale 2018, 10, 11782–11796. [Google Scholar] [CrossRef] [PubMed]
- Telling, N.D.; Everett, J.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Gallagher, J.J.; Wang, J.; Hitchcock, A.P. Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer’s disease. Cell Chem. Biol. 2017, 24, 1205–1215 e3. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Ponce, A.; Collingwood, J.F.; Arellano-Jimenez, M.J.; Zhu, X.; Rogers, J.T.; Betancourt, I.; Jose-Yacaman, M.; Perry, G. High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Sci. Rep. 2016, 6, 24873. [Google Scholar] [CrossRef]
- Collingwood, J.F.; Chong, R.K.; Kasama, T.; Cervera-Gontard, L.; Dunin-Borkowski, R.E.; Perry, G.; Posfai, M.; Siedlak, S.L.; Simpson, E.T.; Smith, M.A.; et al. Three-dimensional tomographic imaging and characterization of iron compounds within Alzheimer’s plaque core material. J. Alzheimers Dis. 2008, 14, 235–245. [Google Scholar] [CrossRef]
- House, E.; Collingwood, J.; Khan, A.; Korchazkina, O.; Berthon, G.; Exley, C. Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer’s disease. J. Alzheimers Dis. 2004, 6, 291–301. [Google Scholar] [CrossRef]
- McLeary, F.A.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Radford, R.A.W.; Okita, Y.; Faller, P.; Chung, R.S.; Pountney, D.L. Switching on Endogenous metal binding proteins in Parkinson’s Disease. Cells 2019, 8, 179. [Google Scholar] [CrossRef]
- Pesch, B.; Casjens, S.; Woitalla, D.; Dharmadhikari, S.; Edmondson, D.A.; Zella, M.A.S.; Lehnert, M.; Lotz, A.; Herrmann, L.; Muhlack, S.; et al. Impairment of motor function correlates with neurometabolite and brain iron alterations in Parkinson’s disease. Cells 2019, 8, 96. [Google Scholar] [CrossRef]
- Mehta, N.J.; Marwah, P.K.; Njus, D. Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin? Cells 2019, 8, 59. [Google Scholar] [CrossRef]
- Li, L.B.; Chai, R.; Zhang, S.; Xu, S.F.; Zhang, Y.H.; Li, H.L.; Fan, Y.G.; Guo, C. Iron Exposure and the Cellular Mechanisms Linked to Neuron Degeneration in Adult Mice. Cells 2019, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of alpha-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.R.; Shin, H.J.; Lee, J.H. Metal-catalyzed oxidation of alpha-synuclein in the presence of Copper(II) and hydrogen peroxide. Arch. Biochem. Biophys. 2000, 378, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova-Golts, N.; Petrucelli, L.; Hardy, J.; Lee, J.M.; Farer, M.; Wolozin, B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 2000, 20, 6048–6054. [Google Scholar] [CrossRef]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Brown, D.R. Alpha-synuclein as a ferrireductase. Biochem. Soc. Trans. 2013, 41, 1513–1517. [Google Scholar] [CrossRef]
- Everett, J.; Cespedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1–42). J. R. Soc. Interface 2014, 11, 20140165. [Google Scholar] [CrossRef]
- Everett, J.; Cespedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Evidence of redox-active iron formation following aggregation of ferrihydrite and the Alzheimer’s disease peptide beta-amyloid. Inorg. Chem. 2014, 53, 2803–2809. [Google Scholar] [CrossRef]
- Lucas, H.R.; Debeer, S.; Hong, M.S.; Lee, J.C. Evidence for copper-dioxygen reactivity during alpha-synuclein fibril formation. J. Am. Chem. Soc. 2010, 132, 6636–6637. [Google Scholar] [CrossRef]
- Khan, A.; Dobson, J.P.; Exley, C. Redox cycling of iron by Abeta42. Free Radic. Biol. Med. 2006, 40, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Collingwood, J.F.; Davidson, M.R. The role of iron in neurodegenerative disorders: Insights and opportunities with synchrotron light. Front. Pharmacol. 2014, 5, 191. [Google Scholar] [CrossRef] [PubMed]
- Chwiej, J.; Adamek, D.; Szczerbowska-Boruchowska, M.; Krygowska-Wajs, A.; Wojcik, S.; Falkenberg, G.; Manka, A.; Lankosz, M. Investigations of differences in iron oxidation state inside single neurons from substantia nigra of Parkinson’s disease and control patients using the micro-XANES technique. J. Biol. Inorg. Chem. 2007, 12, 204–211. [Google Scholar] [CrossRef]
- Collingwood, J.F.; Mikhaylova, A.; Davidson, M.R.; Batich, C.; Streit, W.J.; Terry, J.; Dobson, J. In situ characterization and mapping of iron compounds in Alzheimer’s disease tissue. J. Alzheimer’s Dis. JAD 2005, 7, 267–272. [Google Scholar] [CrossRef]
- Bohic, S.; Murphy, K.; Paulus, W.; Cloetens, P.; Salome, M.; Susini, J.; Double, K. Intracellular chemical imaging of the developmental phases of human neuromelanin using synchrotron X-ray microspectroscopy. Anal. Chem. 2008, 80, 9557–9566. [Google Scholar] [CrossRef]
- Rourk, C.J. Ferritin and neuromelanin “quantum dot” array structures in dopamine neurons of the substantia nigra pars compacta and norepinephrine neurons of the locus coeruleus. Biosystems 2018, 171, 48–58. [Google Scholar] [CrossRef]
- Enochs, W.S.; Sarna, T.; Zecca, L.; Riley, P.A.; Swartz, H.M. The roles of neuromelanin, binding of metal ions, and oxidative cytotoxicity in the pathogenesis of Parkinson’s disease: A hypothesis. J. Neural Transm. -Parkinson’s Dis. Dement. Sect. 1994, 7, 83–100. [Google Scholar] [CrossRef]
- Zecca, L.; Tampellini, D.; Gatti, A.; Crippa, R.; Eisner, M.; Sulzer, D.; Gallorini, M.; Ito, S.; Fariello, R. The neuromelanin of human substantia nigra and its interaction with metals. J. Neural Transm. 2002, 109, 663–672. [Google Scholar] [CrossRef]
- Ortega, R.; Carmona, A.; Llorens, I.; Solari, P.L. X-ray absorption spectroscopy of biological samples. A tutorial. J. Anal. At. Spectrom. 2012, 27, 2054–2056. [Google Scholar] [CrossRef]
- Kiernan, J.A. Histological and Histochemical Methods, 5th ed.; Scion Publishing Ltd.: Banbury, UK, 2015. [Google Scholar]
- Loo, J.A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997, 16, 1–23. [Google Scholar] [CrossRef]
- Benesch, J.L.; Ruotolo, B.T. Mass spectrometry: Come of age for structural and dynamical biology. Curr. Opin. Struct. Biol. 2011, 21, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Leney, A.C.; Heck, A.J. Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lin, T.-Y.; Van Orden, S.L.; Zhao, Y.; Barrow, M.P.; Pizarro, A.M.; Qi, Y.; Sadler, P.J.; O’Connor, P.B. Use of top-down and bottom-up fourier transform ion resonance mass spectrometry for mapping calmodulin modifiedl by platinum anticancer drugs. Anal. Chem. 2011, 83, 9507–9515. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Snelling, J.R.; Barrow, M.P.; Scrivens, J.H.; Sadler, P.J.; O’Connor, P.B. Mass Spectrometric Strategies to Improve the Identification of Pt(II)-Modification Sites on Peptides and Proteins. J. Am. Soc. Mass Spectrom. 2014, 25, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Lermyte, F.; Valkenborg, D.; Loo, J.A.; Sobott, F. Radical solutions: Principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure. Mass Spectrom. Rev. 2018, 37, 750–771. [Google Scholar] [CrossRef]
- Brodbelt, J.S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2016, 88, 30–51. [Google Scholar] [CrossRef]
- Stiving, A.Q.; VanAernum, Z.L.; Busch, F.; Harvey, S.R.; Sarni, S.H.; Wysocki, V.H. Surface-Induced Dissociation: An Effective Method for Characterization of Protein Quaternary Structure. Anal. Chem. 2019, 91, 190–209. [Google Scholar] [CrossRef]
- Wongkongkathep, P.; Han, J.Y.; Choi, T.S.; Yin, S.; Kim, H.I.; Loo, J.A. Native top-down mass spectrometry and ion mobility MS for characterizing the cobalt and manganese metal binding of alpha-synuclein protein. J. Am. Soc. Mass Spectrom. 2018, 29, 1870–1880. [Google Scholar] [CrossRef]
- Loo, J.A.; Hu, P.; Smith, R.D. Interaction of angiotensin peptides and zinc metal ions probed by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 1994, 5, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Nemirovskiy, O.V.; Gross, M.L. Determination of calcium binding sites in gas-phase small peptides by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 1998, 9, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Lermyte, F.; Tsybin, Y.O.; O’Connor, P.B.; Loo, J.A. Top or Middle? Up or Down? Toward a Standard Lexicon for Protein Top-Down and Allied Mass Spectrometry Approaches. J. Am. Soc. Mass Spectrom. 2019, 30, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Corazza, A.; Harvey, I.; Sadler, P.J. 1H,13C-NMR and X-ray absorption studies of copper(I) glutathione complexes. Eur. J. Biochem. 1996, 236, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Alexeev, D.; Hunter, D.J.; Campopiano, D.J.; Sadler, P.J. Oxo-iron clusters in a bacterial iron-trafficking protein: New roles for a conserved motif. Biochem. J. 2003, 376, 35–41. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Kaznacheyev, K.; Osanna, A.; Jacobsen, C.; Plashkevych, O.; Vahtras, O.; Ågren, H.; Carravetta, V.; Hitchcock, A.P. Innershell Absorption Spectroscopy of Amino Acids. J. Phys. Chem. A 2002, 106, 3153–3168. [Google Scholar] [CrossRef]
- Cosmidis, J.; Benzerara, K.; Nassif, N.; Tyliszczak, T.; Bourdelle, F. Characterization of Ca-phosphate biological materials by scanning transmission X-ray microscopy (STXM) at the Ca L2,3-, P L2,3-and C K-edges. Acta Biomater. 2015, 12, 260–269. [Google Scholar] [CrossRef]
- Lermyte, F.; Everett, J.; Lam, Y.P.Y.; Wootton, C.A.; Brooks, J.; Barrow, M.P.; Telling, N.D.; Sadler, P.J.; O’Connor, P.B.; Collingwood, J.F. Metal Ion Binding to the Amyloid β Monomer Studied by Native Top-Down FTICR Mass Spectrometry. J. Am. Soc. Mass Spectrom 2019, 1–12. [Google Scholar] [CrossRef]
- Regan, T.J.; Ohldag, H.; Stamm, C.; Nolting, F.; Luning, J. Chemical effects at metal/oxide interfaces studied by X-ray-absorption spectroscopy. Phys. Rev. B 2001, 64, 214422. [Google Scholar] [CrossRef]
- Carboni, E.; Lingor, P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 2015, 7, 395–404. [Google Scholar] [CrossRef]
- Hureau, C.; Coppel, Y.; Dorlet, P.; Solari, P.L.; Sayen, S.; Guillon, E.; Sabater, L.; Faller, P. Deprotonation of the Asp1-Ala2 peptide bond induces modification of the dynamic copper (II) environment in the amyloid-beta peptide near physiological pH. Angew. Chem. Int. Ed. Engl. 2009, 48, 9522–9525. [Google Scholar] [CrossRef] [PubMed]
- Bousejra-ElGarah, F.; Bijani, C.; Coppel, Y.; Faller, P.; Hureau, C. Iron (II) binding to amyloid-beta, the Alzheimer’s peptide. Inorg. Chem. 2011, 50, 9024–9030. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.G.; Perry, G.; Smith, M.A.; Reddy, V.P. NMR studies of zinc, copper, and iron binding to histidine, the principal metal ion complexing site of amyloid-beta peptide. J. Alzheimers Dis. 2010, 20, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Santo Signorelli, S.; Ferrante, M.; Nicoletti, A.; Zappia, M. Metals and neurodegenerative diseases. A systematic review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef]
- Choi, T.S.; Lee, J.; Han, J.Y.; Jung, B.C.; Wongkongkathep, P.; Loo, J.A.; Lee, M.J.; Kim, H.I. Supramolecular Modulation of Structural Polymorphism in Pathogenic alpha-Synuclein Fibrils Using Copper (II) Coordination. Angew. Chem. Int. Ed. Engl. 2018, 57, 3099–3103. [Google Scholar] [CrossRef]





| Technique | Abbreviation | Description |
|---|---|---|
| Mass Spectrometry | MS | Electrospray ionization (ESI) tandem MS is used to determine regions on peptides where metals bind, by analyzing the molecular masses of fragments where the amide backbone of the peptide has been cleaved while preserving protein-metal interactions. The fragmentation pattern indicates the binding region(s), as the fragments containing the residue(s) that interact(s) with the metal show a characteristic mass increase. |
| Scanning Transmission X-ray Microscopy | STXM | Synchrotron soft X-ray microscopy is used in transmission mode to obtain images at tens of nanometer spatial resolution, acquired sequentially in stacks as a function of energy. These data contain spectral information about the chemistry of each region of interest selected within the image. |
| X-ray Absorption Near-Edge Spectroscopy | XANES | Synchrotron hard X-ray microscopy is used in fluorescence mode to obtain energy scans from elements of interest, where the structure of the spectrum is sensitive to the local chemical environment of the scattering element. |
| Transmission Electron Microscopy | TEM | Electron beam imaging is used to investigate the forms of peptide aggregate present in the samples analyzed by MS, STXM, and XANES. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lermyte, F.; Everett, J.; Brooks, J.; Bellingeri, F.; Billimoria, K.; Sadler, P.J.; O’Connor, P.B.; Telling, N.D.; Collingwood, J.F. Emerging Approaches to Investigate the Influence of Transition Metals in the Proteinopathies. Cells 2019, 8, 1231. https://doi.org/10.3390/cells8101231
Lermyte F, Everett J, Brooks J, Bellingeri F, Billimoria K, Sadler PJ, O’Connor PB, Telling ND, Collingwood JF. Emerging Approaches to Investigate the Influence of Transition Metals in the Proteinopathies. Cells. 2019; 8(10):1231. https://doi.org/10.3390/cells8101231
Chicago/Turabian StyleLermyte, Frederik, James Everett, Jake Brooks, Francesca Bellingeri, Kharmen Billimoria, Peter J. Sadler, Peter B. O’Connor, Neil D. Telling, and Joanna F. Collingwood. 2019. "Emerging Approaches to Investigate the Influence of Transition Metals in the Proteinopathies" Cells 8, no. 10: 1231. https://doi.org/10.3390/cells8101231
APA StyleLermyte, F., Everett, J., Brooks, J., Bellingeri, F., Billimoria, K., Sadler, P. J., O’Connor, P. B., Telling, N. D., & Collingwood, J. F. (2019). Emerging Approaches to Investigate the Influence of Transition Metals in the Proteinopathies. Cells, 8(10), 1231. https://doi.org/10.3390/cells8101231