Intrinsically Disordered SRC-3/AIB1 Protein Undergoes Homeostatic Nuclear Extrusion by Nuclear Budding While Ectopic Expression Induces Nucleophagy

Abstract
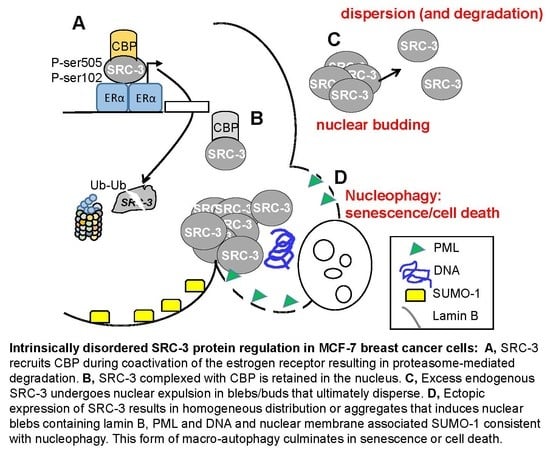
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
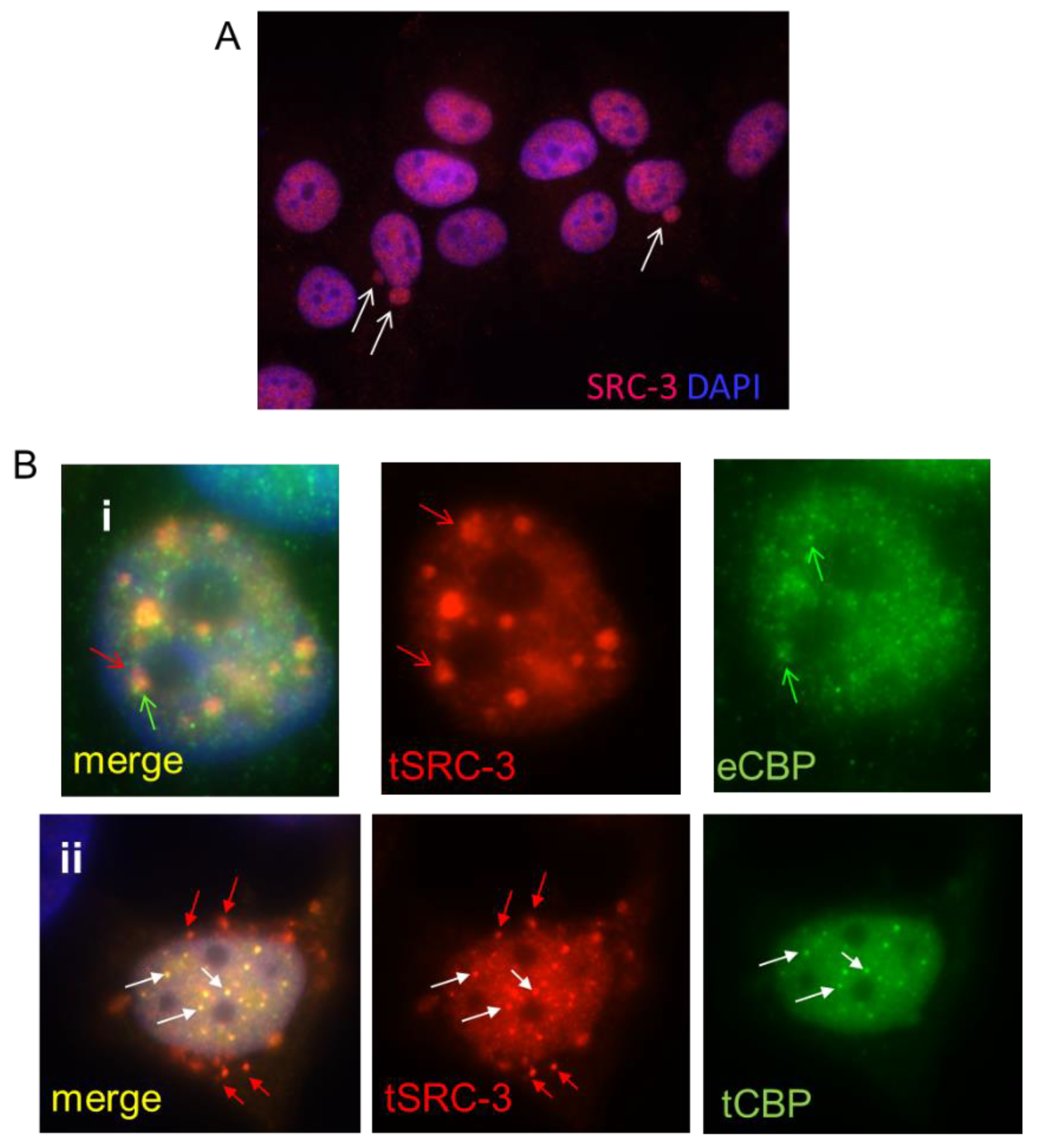
3.1. MCF-7 Cells Extrude Endogenous SRC-3 through Nuclear “Budding” and Altered SRC-3: CBP Stoichiometry Regulates Nuclear Localization and Expulsion: Many Breast Tumors Express Amplified Levels of SRC-3
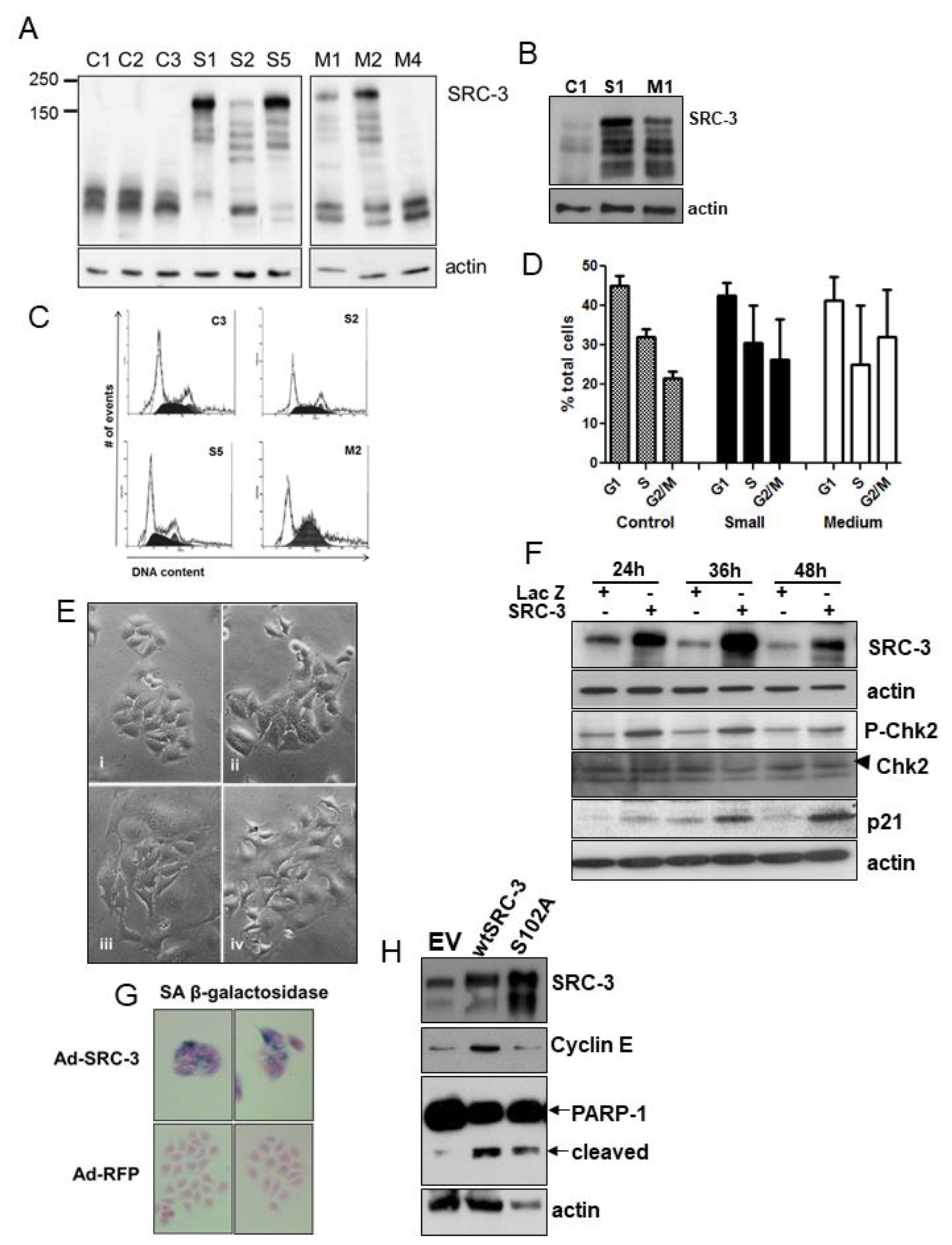
3.2. Overexpressed SRC-3 Induces Cell Cycle Arrest, Senescence, and Apoptosis
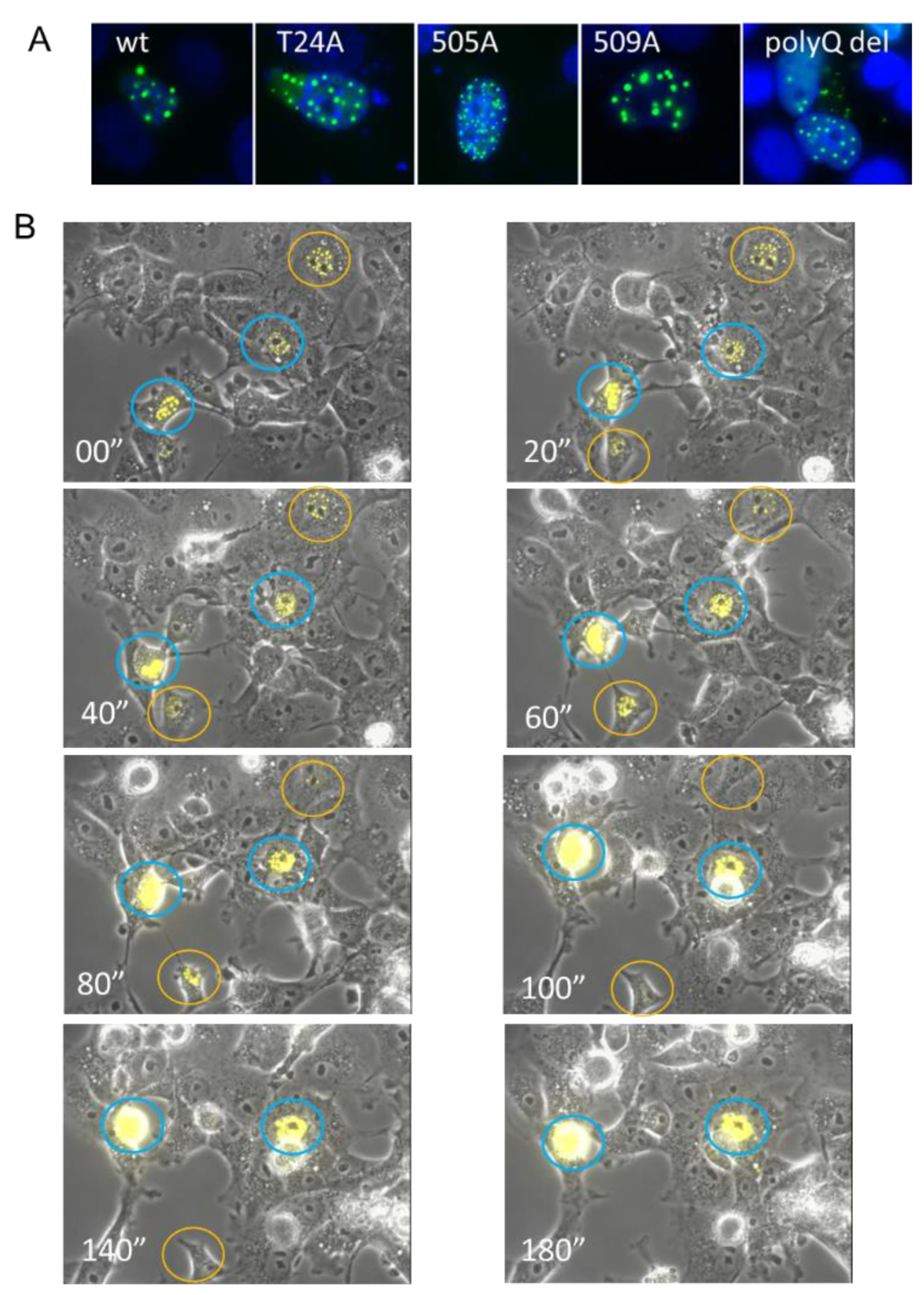
3.3. Ectopically Expressed SRC-3 Protein Forms Nuclear Aggregates
3.4. SRC-3 Overexpression Does Not Affect the Proteasome but Induces Autophagy
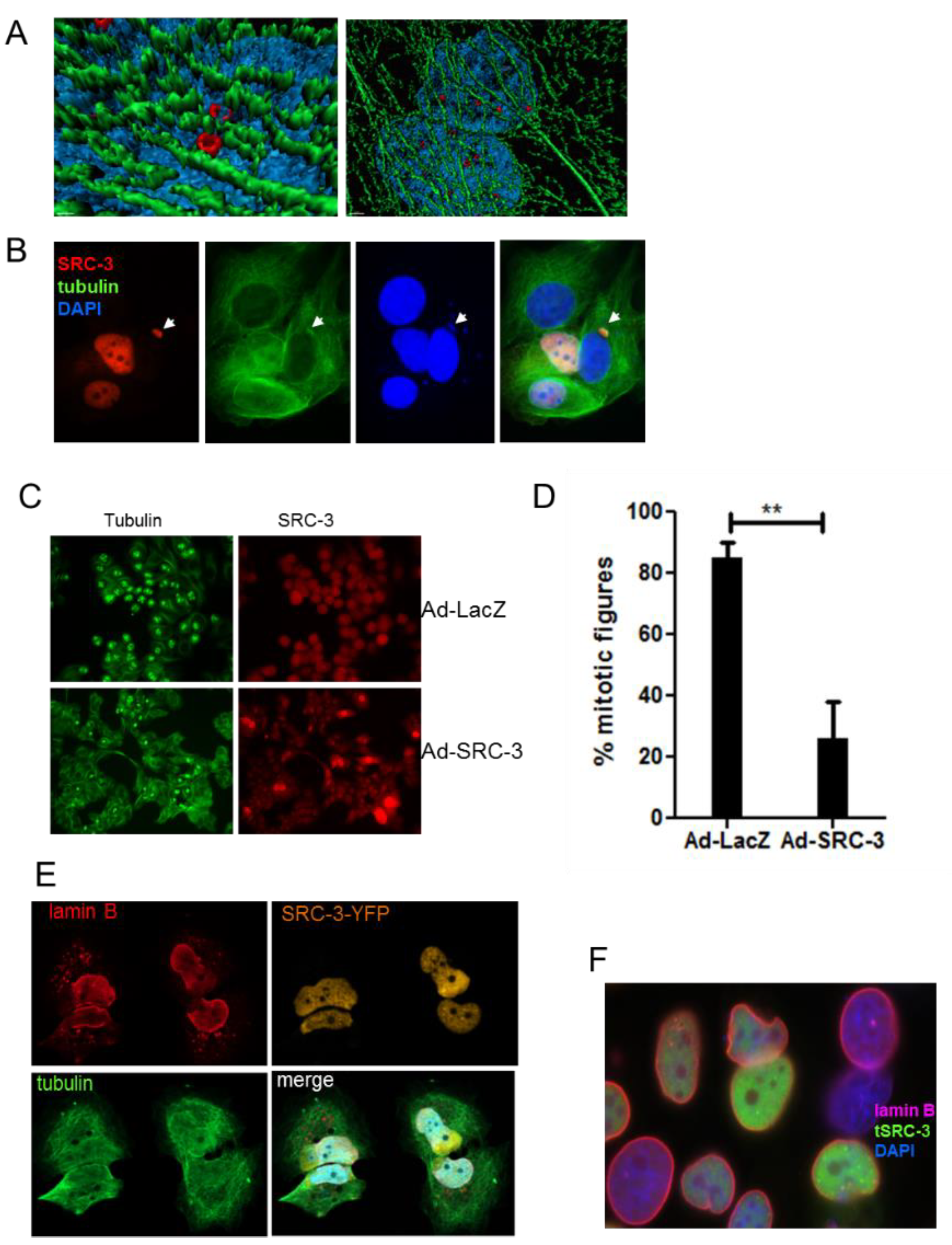
3.5. SRC-3 Nuclear Aggregates Are Proximal to Microtubules and Overexpression Disrupts Both Mitotic Microtubule Dynamics and the Nuclear Membrane
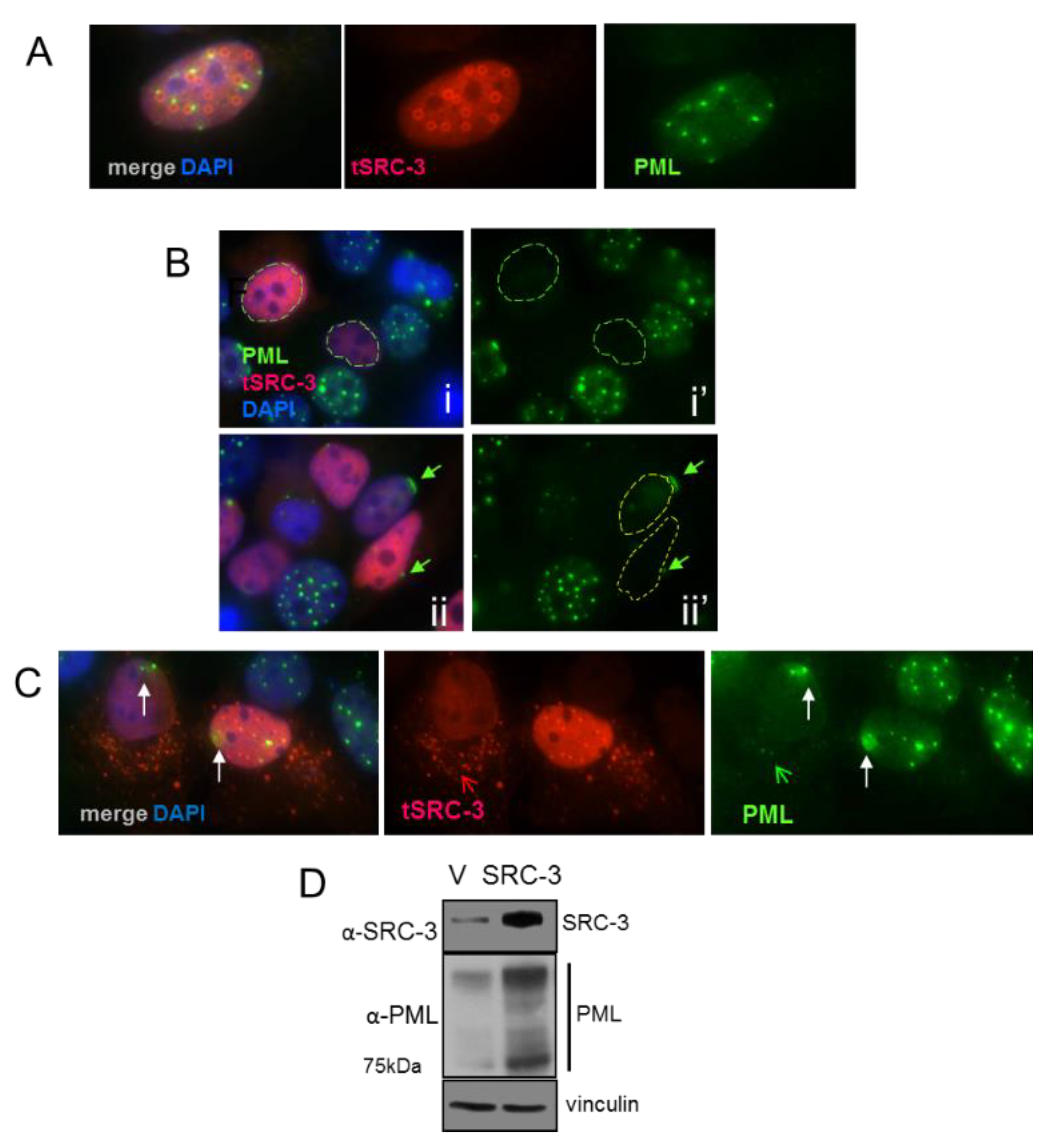
3.6. SRC-3 Overexpression Redistributes PML Bodies and Induces PML
3.7. Increased SRC-3 Protein Level Impacts the SUMO Pathway
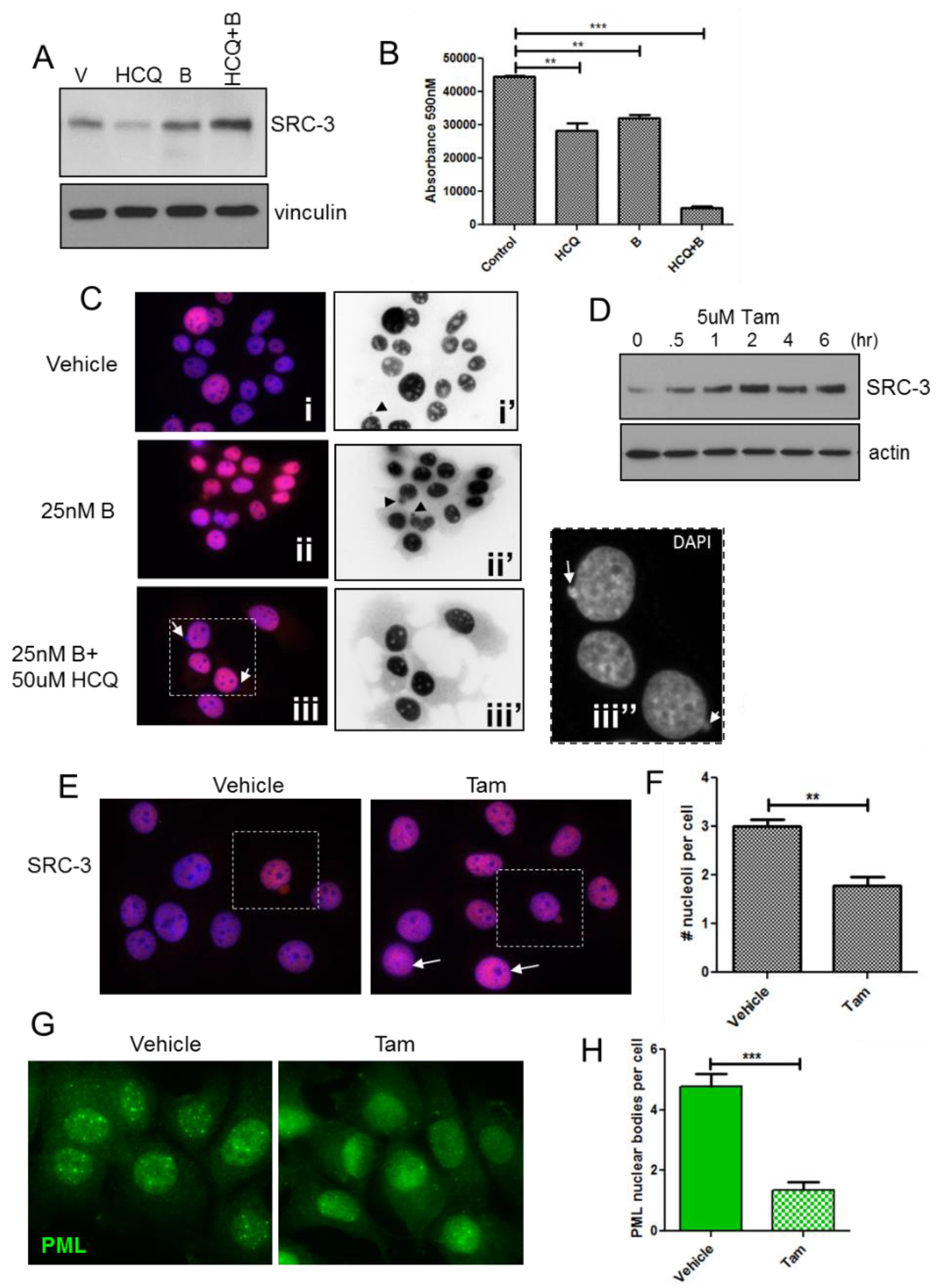
3.8. Pharmacologic Stabilization of SRC-3 Recapitulates Effects of Overexpression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- List, H.J.; Reiter, R.; Singh, B.; Wellstein, A.; Riegel, A.T. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res. Treat. 2001, 68, 21–28. [Google Scholar] [CrossRef]
- Hudelist, G.; Czerwenka, K.; Kubista, E.; Marton, E.; Pischinger, K.; Singer, C.F. Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res. Treat. 2003, 78, 193–204. [Google Scholar] [CrossRef]
- Brown, K.; Chen, Y.; Underhill, T.M.; Mymryk, J.S.; Torchia, J. The coactivator p/CIP/SRC-3 facilitates retinoic acid receptor signaling via recruitment of GCN5. J. Biol. Chem. 2003, 278, 39402–39412. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, R.J.; Schiltz, R.L.; Chakravarti, D.; Nash, A.; Nagy, L.; Privalsky, M.L.; Nakatani, Y.; Evans, R.M. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 1997, 90, 569–580. [Google Scholar] [CrossRef]
- Yan, J.; Yu, C.-T.; Ozen, M.; Ittmann, M.; Tsai, S.Y.; Tsai, M.-J. Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/AKT signaling pathway. Cancer Res. 2006, 66, 11039–11046. [Google Scholar] [CrossRef]
- Xu, J.; Liao, L.; Ning, G.; Yoshida-Komiya, H.; Deng, C.; O’Malley, B.W. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. USA 2000, 97, 6379–6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwase, H.; Omoto, Y.; Toyama, T.; Yamashita, H.; Hara, Y.; Sugiura, H.; Zhang, Z. Clinical significance of AIB1 expression in human breast cancer. Breast Cancer Res. Treat. 2003, 80, 339–345. [Google Scholar] [CrossRef]
- Kirkegaard, T.; McGlynn, L.M.; Campbell, F.M.; Müller, S.; Tovey, S.M.; Dunne, B.; Nielsen, K.V.; Cooke, T.G.; Bartlett, J.M.S. Amplified in breast cancer 1 in human epidermal growth factor receptor-positive tumors of tamoxifen-treated breast cancer patients. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2007, 13, 1405–1411. [Google Scholar] [CrossRef]
- Wu, R.-C.; Feng, Q.; Lonard, D.M.; O’Malley, B.W. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell 2007, 129, 1125–1140. [Google Scholar] [CrossRef]
- Li, C.; Liang, Y.-Y.; Feng, X.-H.; Tsai, S.Y.; Tsai, M.-J.; O’Malley, B.W. Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol. Cell 2008, 31, 835–849. [Google Scholar] [CrossRef]
- Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Shi, B.; Li, R.; Wang, Y.; Liang, J.; Fan, D.; Wu, G.; et al. Coordinated regulation of AIB1 transcriptional activity by sumoylation and phosphorylation. J. Biol. Chem. 2006, 281, 21848–21856. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, C.; Hong, Y.; Bi, H.; Zhao, F.; Liu, Y.; Ao, X.; Pang, P.; Xing, X.; Chang, A.K.; et al. The transcriptional activity of co-activator AIB1 is regulated by the SUMO E3 ligase PIAS1. Biol. Cell 2012, 104, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Martinez-Yamout, M.; Chung, J.; Chen, H.; Xu, W.; Dyson, H.J.; Evans, R.M.; Wright, P.E. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 2002, 415, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.-O.; Bae, S.-H.; Dyson, H.J.; Wright, P.E. NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry 2008, 47, 1299–1308. [Google Scholar] [CrossRef]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Gao, Y.-S.; Sztul, E. Hassles with taking out the garbage: Aggravating aggresomes. Traffic 2002, 3, 388–396. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R. Substrate access and processing by the 20S proteasome core particle. Int. J. Biochem. Cell Biol. 2003, 35, 606–616. [Google Scholar] [CrossRef]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef]
- Bence, N.F.; Bennett, E.J.; Kopito, R.R. Application and analysis of the GFPu family of ubiquitin-proteasome system reporters. Methods Enzymol. 2005, 399, 481–490. [Google Scholar]
- Li, H.; Gomes, P.J.; Chen, J.D. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc. Natl. Acad. Sci. USA 1997, 94, 8479–8484. [Google Scholar] [CrossRef] [Green Version]
- Doucet, J.P.; Murphy, B.J.; Tuana, B.S. Modification of a discontinuous and highly porous sodium dodecyl sulfate-polyacrylamide gel system for minigel electrophoresis. Anal. Biochem. 1990, 190, 209–211. [Google Scholar] [CrossRef]
- Ruddy, S.C.; Lau, R.; Cabrita, M.A.; McGregor, C.; McKay, B.C.; Murphy, L.C.; Wright, J.S.; Durst, T.; Pratt, M.A.C. Preferential estrogen receptor β ligands reduce Bcl-2 expression in hormone-resistant breast cancer cells to increase autophagy. Mol. Cancer Ther. 2014, 13, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Louie, M.C.; Zou, J.X.; Rabinovich, A.; Chen, H.-W. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol. Cell. Biol. 2004, 24, 5157–5171. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Wong, L.-J.C.; Dai, P.; Lu, J.-F.; Lou, M.A.; Clarke, R.; Nazarov, V. AIB1 gene amplification and the instability of polyQ encoding sequence in breast cancer cell lines. BMC Cancer 2006, 6, 111. [Google Scholar] [CrossRef]
- Thenot, S.; Charpin, M.; Bonnet, S.; Cavailles, V. Estrogen receptor cofactors expression in breast and endometrial human cancer cells. Mol. Cell. Endocrinol. 1999, 156, 85–93. [Google Scholar] [CrossRef]
- Azorsa, D.O.; Cunliffe, H.E.; Meltzer, P.S. Association of steroid receptor coactivator AIB1 with estrogen receptor-alpha in breast cancer cells. Breast Cancer Res. Treat. 2001, 70, 89–101. [Google Scholar] [CrossRef]
- Zhou, G.; Hashimoto, Y.; Kwak, I.; Tsai, S.Y.; Tsai, M.-J. Role of the steroid receptor coactivator SRC-3 in cell growth. Mol. Cell. Biol. 2003, 23, 7742–7755. [Google Scholar] [CrossRef]
- Fu, L.; Gao, Y.-S.; Tousson, A.; Shah, A.; Chen, T.-L.L.; Vertel, B.M.; Sztul, E. Nuclear aggresomes form by fusion of PML-associated aggregates. Mol. Biol. Cell 2005, 16, 4905–4917. [Google Scholar] [CrossRef]
- Brünner, N.; Boysen, B.; Jirus, S.; Skaar, T.C.; Holst-Hansen, C.; Lippman, J.; Frandsen, T.; Spang-Thomsen, M.; Fuqua, S.A.; Clarke, R. MCF7/LCC9: An antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997, 57, 3486–3493. [Google Scholar]
- Williams, B.L.; Lipkin, W.I. Endoplasmic reticulum stress and neurodegeneration in rats neonatally infected with borna disease virus. J. Virol. 2006, 80, 8613–8626. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, T.; Simmen, T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.-A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; de Stanchina, E.; Querido, E.; Baptiste, N.; Prives, C.; Lowe, S.W. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000, 14, 2015–2027. [Google Scholar] [PubMed]
- Wolyniec, K.; Shortt, J.; de Stanchina, E.; Levav-Cohen, Y.; Alsheich-Bartok, O.; Louria-Hayon, I.; Corneille, V.; Kumar, B.; Woods, S.J.; Opat, S.; et al. E6AP ubiquitin ligase regulates PML-induced senescence in Myc-driven lymphomagenesis. Blood 2012, 120, 822–832. [Google Scholar] [CrossRef]
- Liebelt, F.; Vertegaal, A.C.O. Ubiquitin-dependent and independent roles of SUMO in proteostasis. Am. J. Physiol. Cell Physiol. 2016, 311, C284–C296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanschitz, L.; De Thé, H.; Le Bras, M. PML, SUMOylation, and Senescence. Front. Oncol. 2013, 3, 171. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.H.; Lin, H.-K.; Scaglioni, P.P.; Yung, T.M.; Pandolfi, P.P. The Mechanisms of PML-Nuclear Body Formation. Mol. Cell 2006, 24, 805. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef]
- Anckar, J.; Sistonen, L. SUMO: Getting it on. Biochem. Soc. Trans. 2007, 35, 1409–1413. [Google Scholar] [CrossRef]
- Nacerddine, K.; Lehembre, F.; Bhaumik, M.; Artus, J.; Cohen-Tannoudji, M.; Babinet, C.; Pandolfi, P.P.; Dejean, A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 2005, 9, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, X.; Zhang, Y.; Gao, Z.; Liu, Y.; Hu, J.; Hu, X.; Li, L.; Shi, J.; Gao, N. Nuclear accumulation of UBC9 contributes to SUMOylation of lamin A/C and nucleophagy in response to DNA damage. J. Exp. Clin. Cancer Res. 2019, 38, 67. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, T.; Das, H.; Hofhaus, G.; Schröder, R.R.; Flotho, A.; Melchior, F. The RanBP2/RanGAP1*SUMO1/Ubc9 SUMO E3 ligase is a disassembly machine for Crm1-dependent nuclear export complexes. Nat. Commun. 2016, 7, 11482. [Google Scholar] [CrossRef] [PubMed]
- Matunis, M.J.; Wu, J.; Blobel, G. SUMO-1 modification and its role in targeting the Ran GTPase-activating protein, RanGAP1, to the nuclear pore complex. J. Cell Biol. 1998, 140, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Yan, J.; Li, R.; Ding, Y.; Thompson, T.C.; Mims, M.P.; Hayes, T.G.; MacDonnell, V.; Lynch, R.G.; Frolov, A.; et al. Bortezomib-mediated inhibition of steroid receptor coactivator-3 degradation leads to activated Akt. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7511–7518. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Gonzalez-Angulo, A.M.; Reuben, J.M.; Booser, D.J.; Pusztai, L.; Krishnamurthy, S.; Esseltine, D.; Stec, J.; Broglio, K.R.; Islam, R.; et al. Bortezomib (VELCADE) in metastatic breast cancer: Pharmacodynamics, biological effects, and prediction of clinical benefits. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2006, 17, 813–817. [Google Scholar] [CrossRef]
- Engel, R.H.; Brown, J.A.; Von Roenn, J.H.; O’Regan, R.M.; Bergan, R.; Badve, S.; Rademaker, A.; Gradishar, W.J. A phase II study of single agent bortezomib in patients with metastatic breast cancer: A single institution experience. Cancer Investig. 2007, 25, 733–737. [Google Scholar] [CrossRef]
- Sun, W.-L.; Chen, J.; Wang, Y.-P.; Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Trinh, X.B.; Sas, L.; Van Laere, S.J.; Prové, A.; Deleu, I.; Rasschaert, M.; Van de Velde, H.; Vinken, P.; Vermeulen, P.B.; Van Dam, P.A.; et al. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncol. Rep. 2012, 27, 657–663. [Google Scholar]
- Wu, W.K.K.; Sakamoto, K.M.; Milani, M.; Aldana-Masankgay, G.; Fan, D.; Wu, K.; Lee, C.W.; Cho, C.H.; Yu, J.; Sung, J.J.Y. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist. Updat. 2010, 13, 87–92. [Google Scholar] [CrossRef]
- Wang, X.J.; Yu, J.; Wong, S.H.; Cheng, A.S.L.; Chan, F.K.L.; Ng, S.S.M.; Cho, C.H.; Sung, J.J.Y.; Wu, W.K.K. A novel crosstalk between two major protein degradation systems: Regulation of proteasomal activity by autophagy. Autophagy 2013, 9, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Lonard, D.M.; Tsai, S.Y.; O’Malley, B.W. Selective estrogen receptor modulators 4-hydroxytamoxifen and raloxifene impact the stability and function of SRC-1 and SRC-3 coactivator proteins. Mol. Cell. Biol. 2004, 24, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, R.; Miller, K.R.; Maiorano, J.N.; Termuhlen, P.M.; Gao, Y.; Berberich, S.J. Novel senescence associated gene, YPEL3, is repressed by estrogen in ER+ mammary tumor cells and required for tamoxifen-induced cellular senescence. Int. J. Cancer 2012, 130, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Anzick, S.L.; Kononen, J.; Walker, R.L.; Azorsa, D.O.; Tanner, M.M.; Guan, X.Y.; Sauter, G.; Kallioniemi, O.P.; Trent, J.M.; Meltzer, P.S. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997, 277, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Qutob, M.S.; Bhattacharjee, R.N.; Pollari, E.; Yee, S.P.; Torchia, J. Microtubule-dependent subcellular redistribution of the transcriptional coactivator p/CIP. Mol. Cell. Biol. 2002, 22, 6611–6626. [Google Scholar] [CrossRef] [PubMed]
- Anzick, S.L.; Azorsa, D.O.; Simons, S.S.; Meltzer, P.S. Phenotypic alterations in breast cancer cells overexpressing the nuclear receptor co-activator AIB1. BMC Cancer 2003, 3, 22. [Google Scholar] [CrossRef]
- Papandreou, M.-E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef]
- Park, Y.-E.; Hayashi, Y.K.; Bonne, G.; Arimura, T.; Noguchi, S.; Nonaka, I.; Nishino, I. Autophagic degradation of nuclear components in mammalian cells. Autophagy 2009, 5, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.; Schlieker, C. Alternative nuclear transport for cellular protein quality control. Trends Cell Biol. 2012, 22, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Kvam, E.; Goldfarb, D.S. Nucleus-vacuole junctions and piecemeal microautophagy of the nucleus in S. cerevisiae. Autophagy 2007, 3, 85–92. [Google Scholar] [CrossRef]
- Shibata, Y.; Morimoto, R.I. How the nucleus copes with proteotoxic stress. Curr. Biol. 2014, 24, R463–R474. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, E.; Sharma, P.; Lockett, S.J.; Lualdi, M.; Kuehn, M.R. Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. J. Cell. Sci. 2008, 121, 4106–4113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jul-Larsen, A.; Grudic, A.; Bjerkvig, R.; Bøe, S.O. Subcellular distribution of nuclear import-defective isoforms of the promyelocytic leukemia protein. BMC Mol. Biol. 2010, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-F.; Yang, T.; Huang, H.; Liu, L.F.; Hwang, J. Phosphorylation of Ubc9 by Cdk1 enhances SUMOylation activity. PLoS ONE 2012, 7, e34250. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Wu, J.; Wang, L.; Fu, Z. SUMOylation Promotes Nuclear Import and Stabilization of Polo-like Kinase 1 to Support Its Mitotic Function. Cell Rep. 2017, 21, 2147–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Riceberg, J.; Soucy, T.; Koenig, E.; Minissale, J.; Gallery, M.; Bernard, H.; Yang, X.; Liao, H.; Rabino, C.; et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 2017, 13, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Dasso, M. The SUMO Pathway in Mitosis. Adv. Exp. Med. Biol. 2017, 963, 171–184. [Google Scholar]
- García-Mata, R.; Bebök, Z.; Sorscher, E.J.; Sztul, E.S. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 1999, 146, 1239–1254. [Google Scholar] [CrossRef]
- Rothballer, A.; Kutay, U. The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma 2013, 122, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.K.; Umeshima, H.; Kurisu, J.; Kengaku, M. Nesprins and opposing microtubule motors generate a point force that drives directional nuclear motion in migrating neurons. Development 2018, 145, 5. [Google Scholar] [CrossRef]
- Mellad, J.A.; Warren, D.T.; Shanahan, C.M. Nesprins LINC the nucleus and cytoskeleton. Curr. Opin. Cell Biol. 2011, 23, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Ahmed, K.; Ding, H.; Ding, X.; Lan, J.; Yang, Z.; Miao, Y.; Zhu, Y.; Shi, Y.; Zhu, J.; et al. Stabilization of PML nuclear localization by conjugation and oligomerization of SUMO-3. Oncogene 2005, 24, 5401–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrita, M.A.; Renart, L.I.; Lau, R.; Pratt, M.A.C. Intrinsically Disordered SRC-3/AIB1 Protein Undergoes Homeostatic Nuclear Extrusion by Nuclear Budding While Ectopic Expression Induces Nucleophagy. Cells 2019, 8, 1278. https://doi.org/10.3390/cells8101278
Cabrita MA, Renart LI, Lau R, Pratt MAC. Intrinsically Disordered SRC-3/AIB1 Protein Undergoes Homeostatic Nuclear Extrusion by Nuclear Budding While Ectopic Expression Induces Nucleophagy. Cells. 2019; 8(10):1278. https://doi.org/10.3390/cells8101278
Chicago/Turabian StyleCabrita, Miguel A., L. Isabel Renart, Rosanna Lau, and M. A. Christine Pratt. 2019. "Intrinsically Disordered SRC-3/AIB1 Protein Undergoes Homeostatic Nuclear Extrusion by Nuclear Budding While Ectopic Expression Induces Nucleophagy" Cells 8, no. 10: 1278. https://doi.org/10.3390/cells8101278
APA StyleCabrita, M. A., Renart, L. I., Lau, R., & Pratt, M. A. C. (2019). Intrinsically Disordered SRC-3/AIB1 Protein Undergoes Homeostatic Nuclear Extrusion by Nuclear Budding While Ectopic Expression Induces Nucleophagy. Cells, 8(10), 1278. https://doi.org/10.3390/cells8101278