Conditional Reprogramming for Patient-Derived Cancer Models and Next-Generation Living Biobanks

 , ,
, ,
Abstract
:1. Traditional Cancer Cell Lines and Animal Cancer Models
2. Genomics-Based Targeting Therapies
3. Patient-Derived Cancer Models and CRC are Needed for Precision Oncology
3.1. iPS (Induce Pluripotent Stem) Cells
3.2. Organoid Cultures
3.3. Patient-Derived Xenografts (PDX)
3.4. Conditionally Reprogrammed Cells (CRCs)
4. An Example of the Matched Normal and TSCC Cancer Model using Conditional Cell Reprogramming (CR) Technology
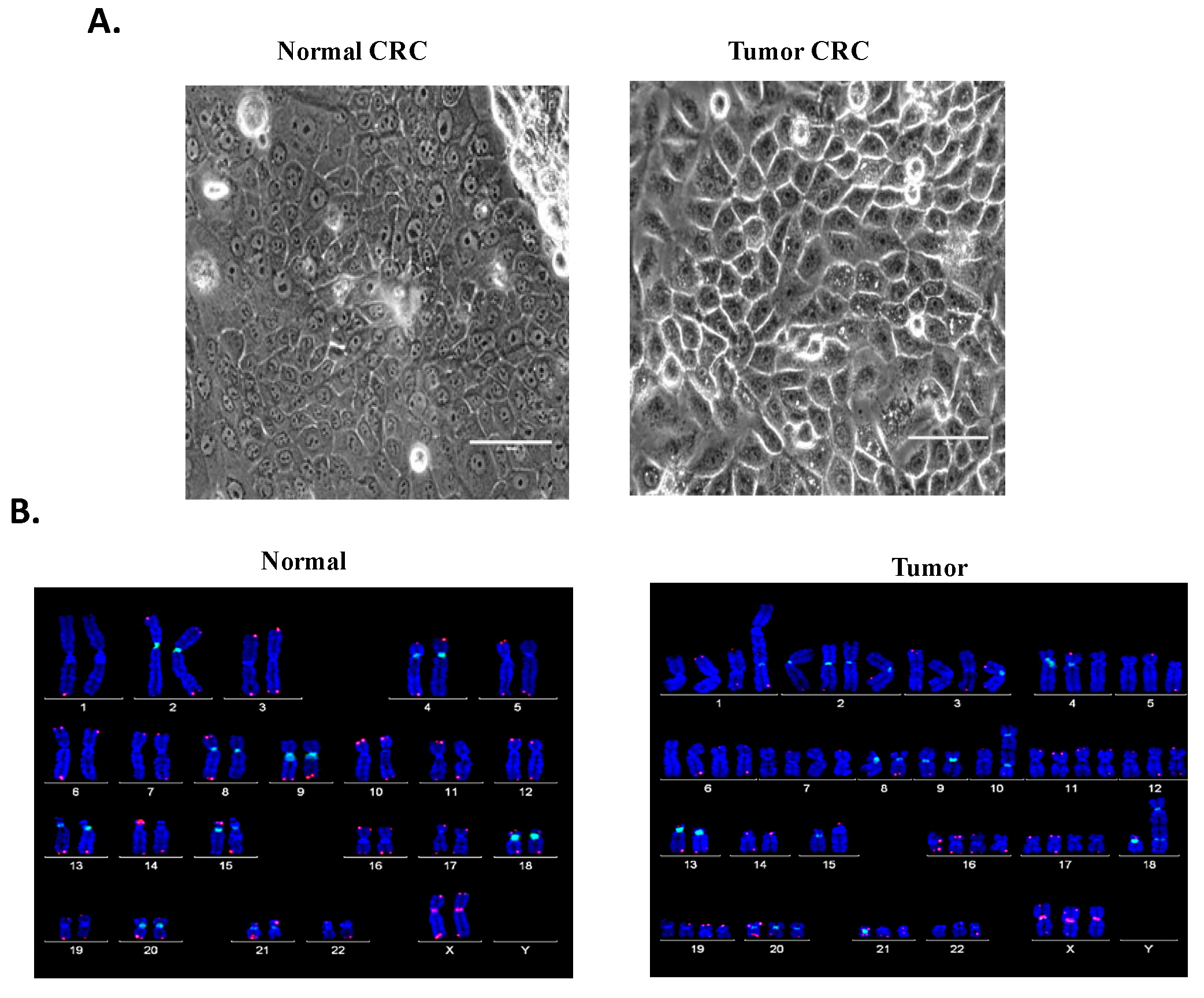
4.1. Establishment of Patient-Derived Matched Normal and TSCC CRCs
4.2. Biological Characterization
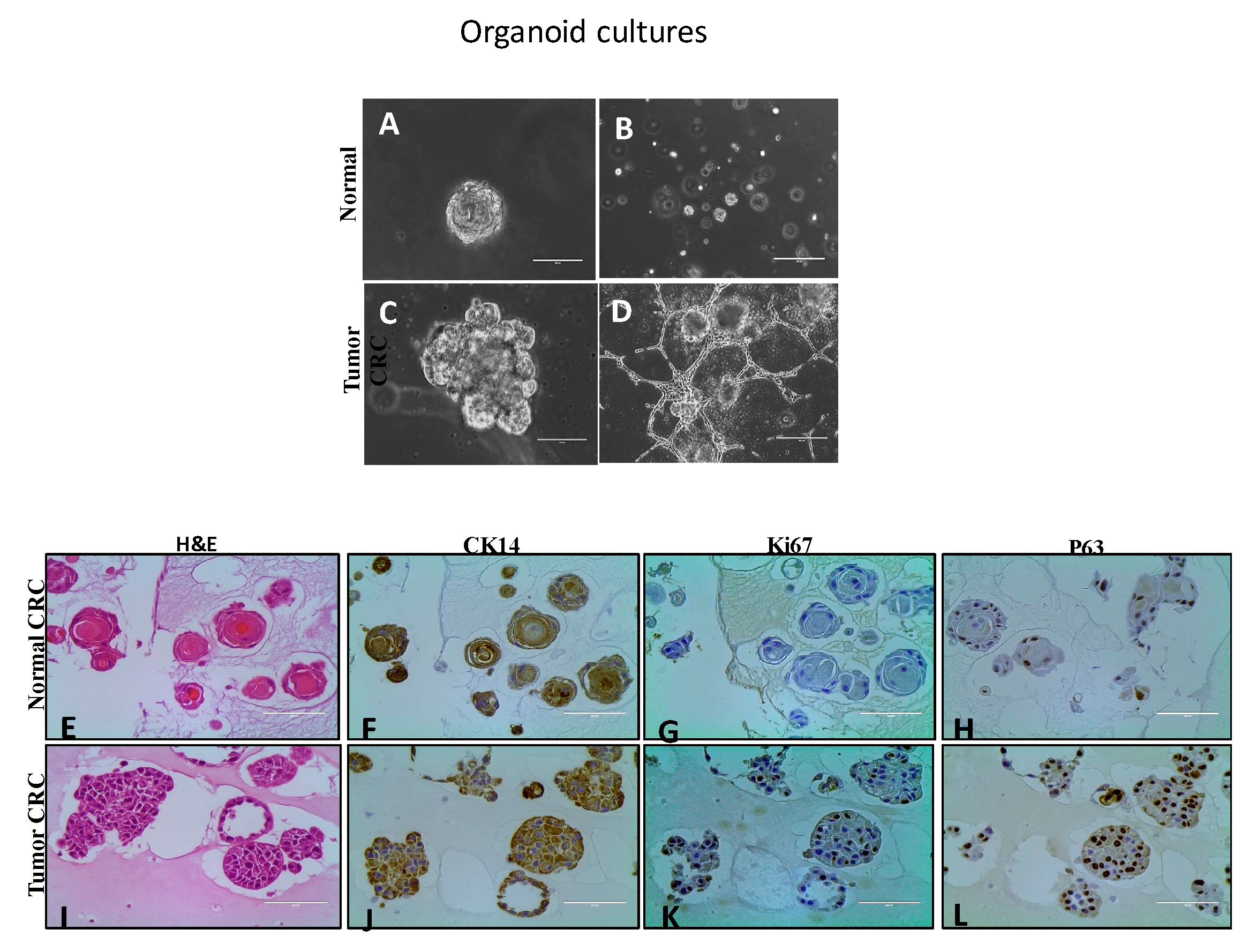
4.2.1. Organoid Cultures
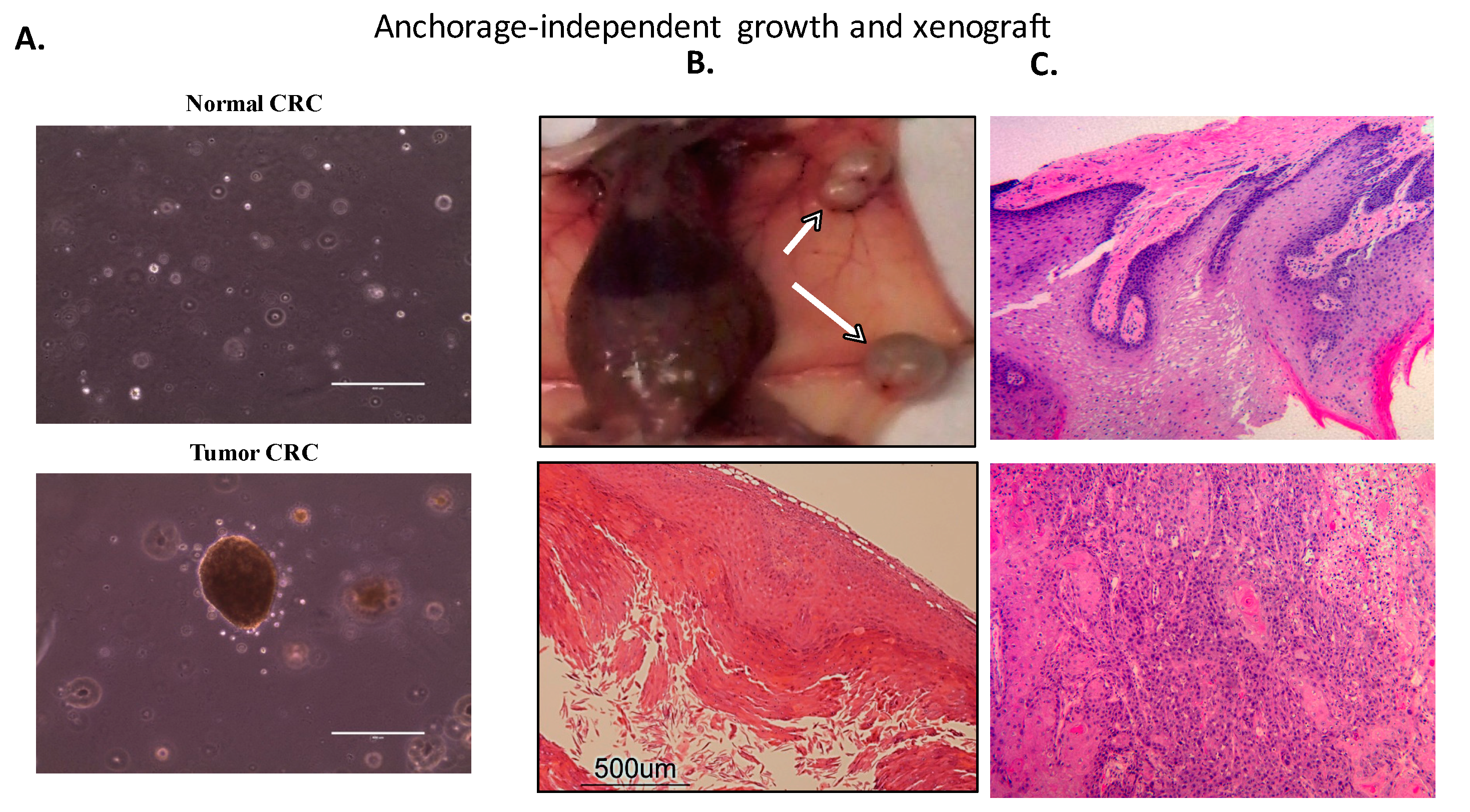
4.2.2. Transformation and Tumorigenicity Assays
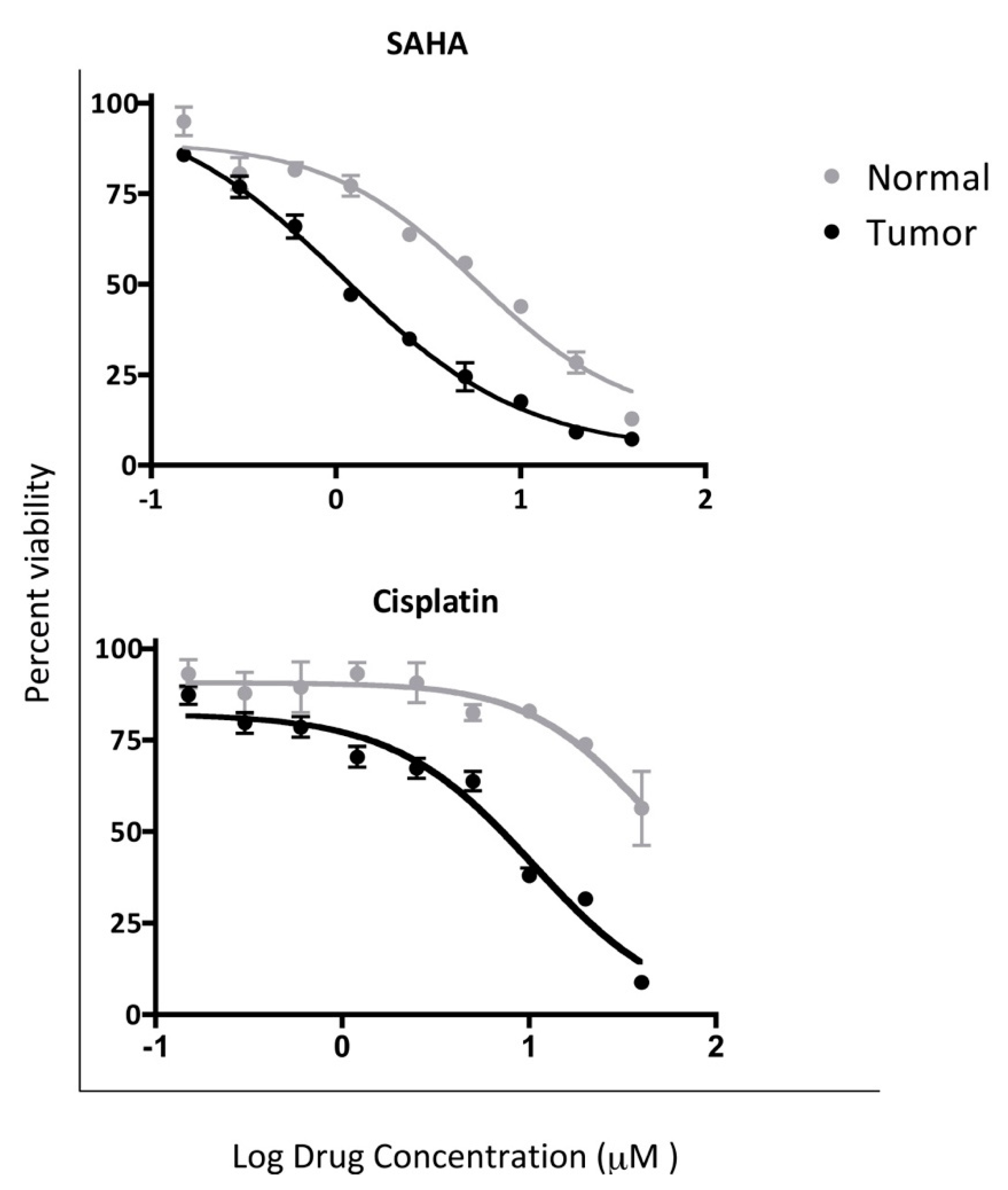
4.3. In Vitro Chemosensitivity of Matched CRCs
4.4. Top Active Pathways in the Tumor CRC Cells
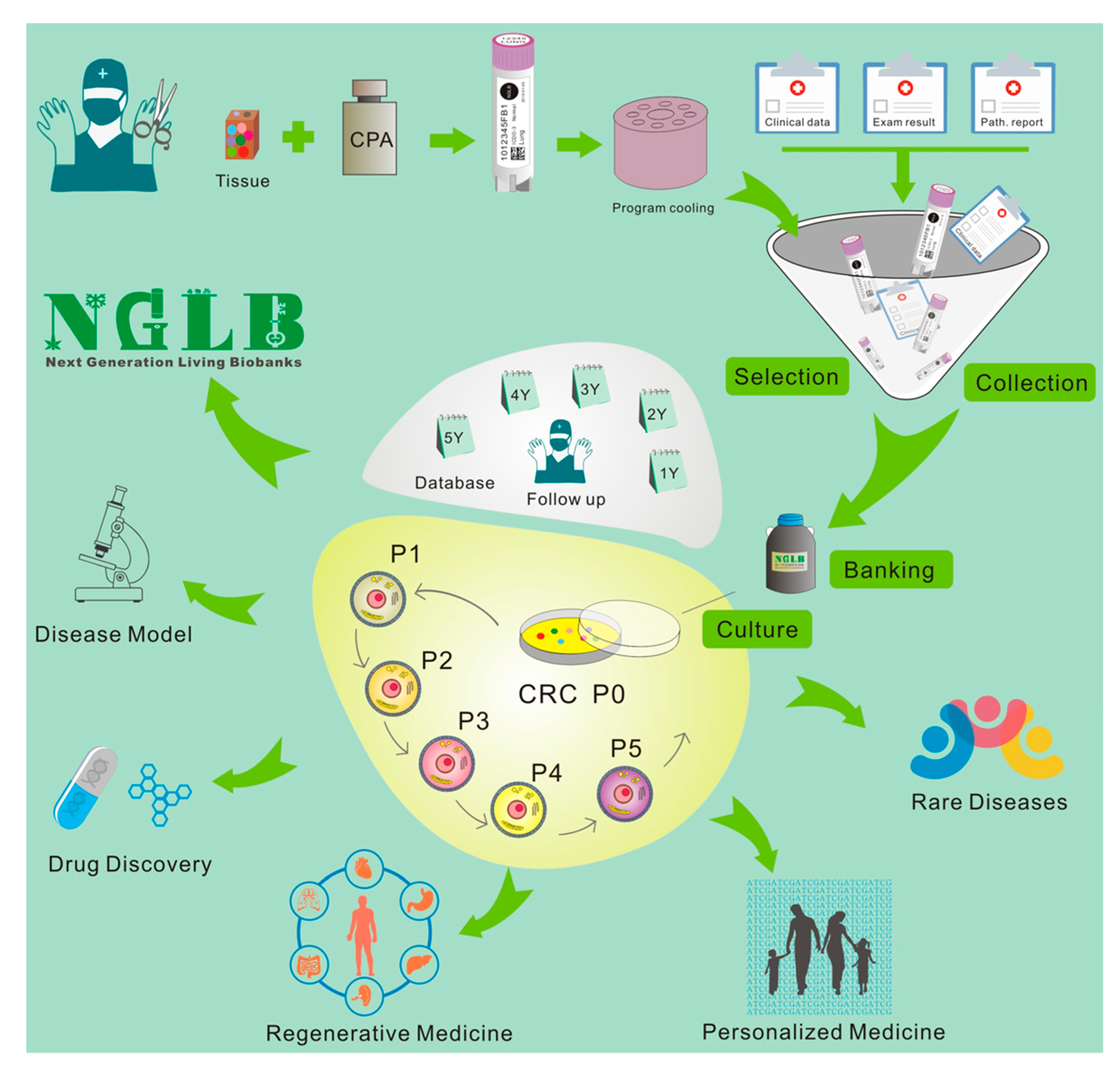
5. Next-Generation Living Biobanks (NGLB)
6. Applications of CR-based NGLB
6.1. Precision Medicine
6.2. Regenerative Medicine
6.3. Modeling Diseases
6.4. Discovery of Novel Targets and Drugs
6.5. Others
Funding
Acknowledgments
Conflicts of Interest
References
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef]
- McDermott, U.; Sharma, S.V.; Dowell, L.; Greninger, P.; Montagut, C.; Lamb, J.; Archibald, H.; Raudales, R.; Tam, A.; Lee, D.; et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. USA 2007, 104, 19936–19941. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Meir, J.; Bedi, A.; Wysocki, P.T.; Hoque, M.O.; Sidransky, D. Patient-derived xenografts as tools in pharmaceutical development. Clin. Pharm. 2016, 99, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dakic, A.; Chen, R.; Disbrow, G.L.; Zhang, Y.; Dai, Y.; Schlegel, R. Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc. J. Virol. 2008, 82, 11568–11576. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Disbrow, G.L.; Yuan, H.; Tomaic, V.; Schlegel, R. Myc and human papillomavirus type 16 E7 genes cooperate to immortalize human keratinocytes. J. Virol. 2007, 81, 12689–12695. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yuan, H.; Fu, B.; Disbrow, G.L.; Apolinario, T.; Tomaic, V.; Kelley, M.L.; Baker, C.C.; Huibregtse, J.; Schlegel, R. The E6AP ubiquitin ligase is required for transactivation of the hTERT promoter by the human papillomavirus E6 oncoprotein. J. Biol. Chem. 2005, 280, 10807–10816. [Google Scholar] [CrossRef]
- Ghittoni, R.; Accardi, R.; Chiocca, S.; Tommasino, M. Role of human papillomaviruses in carcinogenesis. Ecancermedicalscience 2015, 9, 526. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [Green Version]
- Cid Arregui, A.; Gariglio, P.; Kanda, T.; Doorbar, J. ONCOGENIC HUMAN PAPILLOMAVIRUSES: High-Risk Human Papillomaviruses: Towards a Better Understanding of the Mechanisms of Viral Transformation, Latency and Immune-Escape. Open Virol. J. 2012, 6, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Reviews. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H.; Eastern Cooperative Oncology, G. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. New Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. New Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Wen, S.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Aldape, K.; et al. Personalized medicine for patients with advanced cancer in the phase I program at MD anderson: Validation and landmark analyses. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4827–4836. [Google Scholar] [CrossRef]
- Andre, F.; Bachelot, T.; Commo, F.; Campone, M.; Arnedos, M.; Dieras, V.; Lacroix-Triki, M.; Lacroix, L.; Cohen, P.; Gentien, D.; et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: A multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 2014, 15, 267–274. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Canon, J.L.; Braud, F.D.; Grossi, F.; De Pas, T.; Gray, J.E.; Su, W.C.; Felip, E.; Yoshioka, H.; Gridelli, C.; et al. Safety and Efficacy of Buparlisib (BKM120) in Patients with PI3K Pathway-Activated Non-Small Cell Lung Cancer: Results from the Phase II BASALT-1 Study. J. Thorac. Oncol. 2015, 10, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Benefit From Genome-Driven Oncology. Jama. Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Letai, A. Functional precision cancer medicine-moving beyond pure genomics. Nat. Med. 2017, 23, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Leiserson, M.D.M.; Ruppin, E.; Ronai, Z.A. Precision Oncology: The Road Ahead. Trends Mol. Med. 2017, 23, 874–898. [Google Scholar] [CrossRef] [PubMed]
- Curry, E.L.; Moad, M.; Robson, C.N.; Heer, R. Using induced pluripotent stem cells as a tool for modelling carcinogenesis. World J. Stem Cells 2015, 7, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Fukuda, K. Methods of induced pluripotent stem cells for clinical application. World J. Stem Cells 2015, 7, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Marin Navarro, A.; Susanto, E.; Falk, A.; Wilhelm, M. Modeling cancer using patient-derived induced pluripotent stem cells to understand development of childhood malignancies. Cell Death Discov. 2018, 4, 7. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Reviews. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef]
- Papaetrou, E.P. Author Correction: Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2019, 25, 861. [Google Scholar] [CrossRef]
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadauld, L.D.; Garcia, S.; Natsoulis, G.; Bell, J.M.; Miotke, L.; Hopmans, E.S.; Xu, H.; Pai, R.K.; Palm, C.; Regan, J.F.; et al. Metastatic tumor evolution and organoid modeling implicate TGFBR2 as a cancer driver in diffuse gastric cancer. Genome Biol. 2014, 15, 428. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Clevers, H. Organoid cultures for the analysis of cancer phenotypes. Curr. Opin. Genet. Dev. 2014, 24, 68–73. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef]
- Ewald, A.J. 3D cell biology—the expanding frontier. J. Cell Sci. 2017, 130, 1. [Google Scholar] [CrossRef]
- Shamir, E.R.; Ewald, A.J. Three-dimensional organotypic culture: Experimental models of mammalian biology and disease. Nat. Reviews. Mol. Cell Biol. 2014, 15, 647–664. [Google Scholar] [CrossRef]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid technology and applications in cancer research. J. Hematol. Oncol. 2018, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Artegiani, B.; Clevers, H. Use and application of 3D-organoid technology. Hum. Mol. Genet. 2018, 27, R99–R107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef]
- Xinaris, C. Organoids for replacement therapy: Expectations, limitations and reality. Curr. Opin. Organ. Transpl. 2019, 24, 555–561. [Google Scholar] [CrossRef]
- Jin, K.; Teng, L.; Shen, Y.; He, K.; Xu, Z.; Li, G. Patient-derived human tumour tissue xenografts in immunodeficient mice: A systematic review. Clin. Transl. Oncol.: Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2010, 12, 473–480. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Reviews. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.A.; Anderson, W.C.; Santaguida, M.T.; Dylla, S.J. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab. Investig. 2013, 93, 970–982. [Google Scholar] [CrossRef]
- Suprynowicz, F.A.; Upadhyay, G.; Krawczyk, E.; Kramer, S.C.; Hebert, J.D.; Liu, X.; Yuan, H.; Cheluvaraju, C.; Clapp, P.W.; Boucher, R.C., Jr.; et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc. Natl. Acad. Sci. USA. 2012, 109, 20035–20040. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.-L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439. [Google Scholar] [CrossRef] [PubMed]
- Alamri, A.M.; Kang, K.; Groeneveld, S.; Wang, W.; Zhong, X.; Kallakury, B.; Hennighausen, L.; Liu, X.; Furth, P.A. Primary cancer cell culture: Mammary-optimized vs conditional reprogramming. Endocr. Relat. Cancer 2016, 23, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Gardell, A.M.; Qin, Q.; Rice, R.H.; Li, J.; Kültz, D. Derivation and osmotolerance characterization of three immortalized tilapia (Oreochromis mossambicus) cell lines. PLoS ONE 2014, 9, e95919. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ye, L.; Wei, G.; Chen, Y.; Ye, L.; Wu, X.; Zeng, Z.; Wang, Y.; Yin, G.; Long, X.; et al. Conditional reprogrammed human limbal epithelial cells represent a novel in vitro cell model for drug responses. Biochem. Biophys. Res. Commun. 2018, 499, 735–742. [Google Scholar] [CrossRef]
- Alkhilaiwi, F.; Wang, L.; Zhou, D.; Raudsepp, T.; Ghosh, S.; Paul, S.; Palechor-Ceron, N.; Brandt, S.; Luff, J.; Liu, X.; et al. Long-term expansion of primary equine keratinocytes that maintain the ability to differentiate into stratified epidermis. Stem Cell Res. Ther. 2018, 9, 181. [Google Scholar] [CrossRef]
- O’Malley, Y.; Rotti, P.G.; Thornell, I.M.; Vanegas Calderón, O.G.; Febres-Aldana, C.; Durham, K.; Yao, J.; Li, X.; Zhu, Z.; Norris, A.W.; et al. Development of a polarized pancreatic ductular cell epithelium for physiological studies. J. Appl. Physiol. 2018, 125, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Saffari, P.S.; Vapniarsky, N.; Pollack, A.S.; Gong, X.; Vennam, S.; Pollack, A.J.; Verstraete, F.J.M.; West, R.B.; Arzi, B.; Pollack, J.R. Most canine ameloblastomas harbor HRAS mutations, providing a novel large-animal model of RAS-driven cancer. Oncogenesis 2019, 8, 11. [Google Scholar] [CrossRef]
- McAuliffe, P.F.; Evans, K.W.; Akcakanat, A.; Chen, K.; Zheng, X.; Zhao, H.; Eterovic, A.K.; Sangai, T.; Holder, A.M.; Sharma, C.; et al. Ability to Generate Patient-Derived Breast Cancer Xenografts Is Enhanced in Chemoresistant Disease and Predicts Poor Patient Outcomes. PLoS ONE 2015, 10, e0136851. [Google Scholar] [CrossRef]
- Saeed, K.; Rahkama, V.; Eldfors, S.; Bychkov, D.; Mpindi, J.P.; Yadav, B.; Paavolainen, L.; Aittokallio, T.; Heckman, C.; Wennerberg, K.; et al. Comprehensive Drug Testing of Patient-derived Conditionally Reprogrammed Cells from Castration-resistant Prostate Cancer. Eur. Urol. 2016. [Google Scholar] [CrossRef]
- Beglyarova, N.; Banina, E.; Zhou, Y.; Mukhamadeeva, R.; Andrianov, G.; Bobrov, E.; Lysenko, E.; Skobeleva, N.; Gabitova, L.; Restifo, D.; et al. Screening of Conditionally Reprogrammed Patient-Derived Carcinoma Cells Identifies ERCC3-MYC Interactions as a Target in Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 6153–6163. [Google Scholar] [CrossRef]
- Walters, B.J.; Diao, S.; Zheng, F.; Walters, B.J.; Layman, W.S.; Zuo, J. Pseudo-immortalization of postnatal cochlear progenitor cells yields a scalable cell line capable of transcriptionally regulating mature hair cell genes. Sci. Rep. 2015, 5, 17792. [Google Scholar] [CrossRef]
- Brown, D.D.; Dabbs, D.J.; Lee, A.V.; McGuire, K.P.; Ahrendt, G.M.; Bhargava, R.; Davidson, N.E.; Brufsky, A.M.; Johnson, R.R.; Oesterreich, S.; et al. Developing in vitro models of human ductal carcinoma in situ from primary tissue explants. Breast Cancer Res. Treat. 2015, 153, 311–321. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. Ca. Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Silverman, S., Jr. Demographics and occurrence of oral and pharyngeal cancers. The outcomes, the trends, the challenge. J. Am. Dent. Assoc. 2001, 132 (Suppl. 1), 7S–11S. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol 2008, 26, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Gorogh, T.; Quabius, E.S.; Meyer, P.; Hoffmann, M. Characterisation of seven newly established head and neck squamous cell carcinoma cell lines. Eur. Arch. Otorhinolaryngol. 2015, 272, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Palechor-Ceron, N.; Suprynowicz, F.A.; Upadhyay, G.; Dakic, A.; Minas, T.; Simic, V.; Johnson, M.; Albanese, C.; Schlegel, R.; Liu, X. Radiation induces diffusible feeder cell factor(s) that cooperate with ROCK inhibitor to conditionally reprogram and immortalize epithelial cells. Am. J. Pathol. 2013, 183, 1862–1870. [Google Scholar] [CrossRef]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef]
- Pal, A.; Kleer, C.G. Three dimensional cultures: A tool to study normal acinar architecture vs. malignant transformation of breast cells. J. Vis. Exp. 2014. [Google Scholar] [CrossRef]
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef]
- Lin, C.J.; Grandis, J.R.; Carey, T.E.; Gollin, S.M.; Whiteside, T.L.; Koch, W.M.; Ferris, R.L.; Lai, S.Y. Head and neck squamous cell carcinoma cell lines: Established models and rationale for selection. Head Neck 2007, 29, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Bratthauer, G.L.; Saenger, J.S.; Strauss, B.L. Antibodies targeting p63 react specifically in the cytoplasm of breast epithelial cells exhibiting secretory differentiation. Histopathology 2005, 47, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Sneeuwloper, G.; Snow, G.B. The potential of the nude mouse xenograft model for the study of head and neck cancer. Arch. Otorhinolaryngol 1984, 239, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, J.; Mi, W.; Yang, J.; Han, F.; Lu, X.; Yu, W. Silencing of GM3 synthase suppresses lung metastasis of murine breast cancer cells. Breast Cancer Res. 2008, 10, R1. [Google Scholar] [CrossRef] [PubMed]
- Crouch, S.P.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Martin, K.H.; Slack, J.K.; Boerner, S.A.; Martin, C.C.; Parsons, J.T. Integrin connections map: To infinity and beyond. Science 2002, 296, 1652–1653. [Google Scholar] [CrossRef]
- Aguilar-Rojas, A.; Huerta-Reyes, M. Human gonadotropin-releasing hormone receptor-activated cellular functions and signaling pathways in extra-pituitary tissues and cancer cells (Review). Oncol Rep. 2009, 22, 981–990. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Paskal, W.; Paskal, A.M.; Debski, T.; Gryziak, M.; Jaworowski, J. Aspects of Modern Biobank Activity - Comprehensive Review. Pathol. Oncol. Res. 2018, 24, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Giwa, S.; Lewis, J.K.; Alvarez, L.; Langer, R.; Roth, A.E.; Church, G.M.; Markmann, J.F.; Sachs, D.H.; Chandraker, A.; Wertheim, J.A.; et al. The promise of organ and tissue preservation to transform medicine. Nat. Biotechnol. 2017, 35, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Rimm, D.L. Making every cell like HeLa a giant step for cell culture. Am. J. Pathol. 2012, 180, 443–445. [Google Scholar] [CrossRef]
- Lisanti, M.P.; Tanowitz, H.B. Translational discoveries, personalized medicine, and living biobanks of the future. Am. J. Pathol. 2012, 180, 1334–1336. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386 e310. [Google Scholar] [CrossRef]
- Mullenders, J.; de Jongh, E.; Brousali, A.; Roosen, M.; Blom, J.P.A.; Begthel, H.; Korving, J.; Jonges, T.; Kranenburg, O.; Meijer, R.; et al. Mouse and human urothelial cancer organoids: A tool for bladder cancer research. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017. [Google Scholar] [CrossRef]
- Mondal, A.M.; Ma, A.H.; Li, G.; Krawczyk, E.; Yuan, R.; Lu, J.; Schlegel, R.; Stamatakis, L.; Kowalczyk, K.J.; Philips, G.K.; et al. Fidelity of a PDX-CR model for bladder cancer. Biochem. Biophys. Res. Commun. 2019, 517, 49–56. [Google Scholar] [CrossRef]
- Martini, A.; Sfakianos, J.P.; Galsky, M.D. Conditionally Reprogrammed Patient-derived Cells: A Step Forward Towards Personalized Medicine? Eur. Urol. 2019, 76, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, E.; Hong, S.H.; Galli, S.; Trinh, E.; Wietlisbach, L.; Misiukiewicz, S.F.; Tilan, J.U.; Chen, Y.S.; Schlegel, R.; Kitlinska, J. Murine neuroblastoma cell lines developed by conditional reprogramming preserve heterogeneous phenotypes observed in vivo. Lab. Investig. A J. Tech. Methods Pathol. 2019. [Google Scholar] [CrossRef]
- Kettunen, K.; Bostrom, P.J.; Lamminen, T.; Heinosalo, T.; West, G.; Saarinen, I.; Kaipio, K.; Rantala, J.; Albanese, C.; Poutanen, M.; et al. Personalized Drug Sensitivity Screening for Bladder Cancer Using Conditionally Reprogrammed Patient-derived Cells. Eur. Urol. 2019, 76, 430–434. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, J.; Yang, C.; Tan, R.; Hou, J.; Shi, Y.; Zhang, H.; Ma, S.; Wang, J.; Zhang, M.; et al. Continuous culture of urine-derived bladder cancer cells for precision medicine. Protein Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Yip, Y.L.; Lin, W.; Deng, W.; Jia, L.; Lo, K.W.; Busson, P.; Verillaud, B.; Liu, X.; Tsang, C.M.; Lung, M.L.; et al. Establishment of a nasopharyngeal carcinoma cell line capable of undergoing lytic Epstein-Barr virus reactivation. Lab. Investig. 2018, 98, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smith, C.C.; Utsumi, T.; Bixby, L.M.; Kardos, J.; Wobker, S.E.; Stewart, K.G.; Chai, S.; Manocha, U.; Byrd, K.M.; et al. Molecular Subtype-Specific Immunocompetent Models of High-Grade Urothelial Carcinoma Reveal Differential Neoantigen Expression and Response to Immunotherapy. Cancer Res. 2018, 78, 3954–3968. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Qu, Y.; Gomez, L.J.; Chung, S.; Han, B.; Gao, B.; Yue, Y.; Gong, Y.; Liu, X.; Amersi, F.; et al. Characterization of primary human mammary epithelial cells isolated and propagated by conditional reprogrammed cell culture. Oncotarget 2018, 9, 11503–11514. [Google Scholar] [CrossRef] [PubMed]
- Alamri, A.M.; Liu, X.; Blancato, J.K.; Haddad, B.R.; Wang, W.; Zhong, X.; Choudhary, S.; Krawczyk, E.; Kallakury, B.V.; Davidson, B.J.; et al. Expanding primary cells from mucoepidermoid and other salivary gland neoplasms for genetic and chemosensitivity testing. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Yuan, H.; Krawczyk, E.; Blancato, J.; Albanese, C.; Zhou, D.; Wang, N.; Paul, S.; Alkhilaiwi, F.; Palechor-Ceron, N.; Dakic, A.; et al. HPV positive neuroendocrine cervical cancer cells are dependent on Myc but not E6/E7 viral oncogenes. Sci. Rep. 2017, 7, 45617. [Google Scholar] [CrossRef] [Green Version]
- Timofeeva, O.A.; Palechor-Ceron, N.; Li, G.; Yuan, H.; Krawczyk, E.; Zhong, X.; Liu, G.; Upadhyay, G.; Dakic, A.; Yu, S.; et al. Conditionally reprogrammed normal and primary tumor prostate epithelial cells: A novel patient-derived cell model for studies of human prostate cancer. Oncotarget 2017, 8, 22741–22758. [Google Scholar] [CrossRef]
- Suprynowicz, F.A.; Kamonjoh, C.M.; Krawczyk, E.; Agarwal, S.; Wellstein, A.; Agboke, F.A.; Choudhury, S.; Liu, X.; Schlegel, R. Conditional cell reprogramming involves non-canonical beta-catenin activation and mTOR-mediated inactivation of Akt. PLoS ONE 2017, 12, e0180897. [Google Scholar] [CrossRef]
- Mahajan, A.S.; Sugita, B.M.; Duttargi, A.N.; Saenz, F.; Krawczyk, E.; McCutcheon, J.N.; Fonseca, A.S.; Kallakury, B.; Pohlmann, P.; Gusev, Y.; et al. Genomic comparison of early-passage conditionally reprogrammed breast cancer cells to their corresponding primary tumors. PLoS ONE 2017, 12, e0186190. [Google Scholar] [CrossRef]
- Chen, C.; Choudhury, S.; Wangsa, D.; Lescott, C.J.; Wilkins, D.J.; Sripadhan, P.; Liu, X.; Wangsa, D.; Ried, T.; Moskaluk, C.; et al. A multiplex preclinical model for adenoid cystic carcinoma of the salivary gland identifies regorafenib as a potential therapeutic drug. Sci. Rep. 2017, 7, 11410. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, A.; McQuiston, T.J.; Stetson, D.; Ahmed, A.; Whitston, D.; Zhang, J.; Grondine, M.; Lawson, D.; Challberg, S.S.; Zinda, M.; et al. Generation of stable PDX derived cell lines using conditional reprogramming. Mol. Cancer 2017, 16, 177. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Myers, S.; Wang, J.; Zhou, D.; Woo, J.A.; Kallakury, B.; Ju, A.; Bazylewicz, M.; Carter, Y.M.; Albanese, C.; et al. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. N. Engl. J. Med. 2012, 367, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Zhangyan, L.I.; Tang, Y.; Luo, H.; Cheng, C.; Zunfu, K.E.; Chen, X.J.P.T. In Vitro Culture of long-term Patient-derived Conditionally Reprogrammed Cells from Lung Cancer and Application to Individual Chemotherapeutic Susceptibility Studies. Pharm. Today 2017, 2, 113–117. [Google Scholar]
- Boström, P.; Kettunen, K.; Lamminen, T.; Heinosalo, T.; West, G.; Poutanen, M.; Rantala, J.; Taimen, P. 462 - High-throughput drug screening using conditionally reprogrammed patient-derived cell lines in bladder cancer. Eur. Urol. Suppl. 2018, 17, e662. [Google Scholar] [CrossRef]
- Hollevoet, K.; Mason-Osann, E.; Liu, X.F.; Imhof-Jung, S.; Niederfellner, G.; Pastan, I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol. Cancer Ther. 2014, 13, 2040–2049. [Google Scholar] [CrossRef]
- Mimoto, R.; Fushimi, A.; Kazama, T.; Nogi, H.; Takeyama, H. Conditional Reprogramming Cells Are Novel Tools for Drug Response Assay and the Development of Personalized Medicine in Luminal-B Breast Cancer. J. Am. Coll. Surg. 2018, 227, e79. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Y.; Xiao, Y. Re: Kimmo Kettunen, Peter J. Bostrom, Tarja Lamminen; et al. Personalized Drug Sensitivity Screening for Bladder Cancer Using Conditionally Reprogrammed Patient-derived Cells. Eur Urol. In press. Eur. Urol. 2019. [Google Scholar] [CrossRef]
- Kettunen, K.; Bostrom, P.; Taimen, P. Reply to Xuefeng Liu’s Letter to the Editor, re: Kimmo Kettunen, Peter J. Bostrom, Tarja Lamminen; et al. Personalized Drug Sensitivity Screening for Bladder Cancer Using Conditionally Reprogrammed Patient-derived Cells. Eur Urol. In press. https://doi.org/10.1016/j.eururo.2019.06.016: Can Patient-derived Cancer Models Change the Costliest Cancer Type? Eur. Urol. 2019. [Google Scholar] [CrossRef]
- Kettunen, K.; Bostrom, P.J.; Taimen, P. Reply to Mengxin Lu, Yi Zhang, Yu Xiao’s Letter to the Editor, re: Kimmo Kettunen, Peter J. Bostrom, Tarja Lamminen; et al. Personalized Drug Sensitivity Screening for Bladder Cancer Using Conditionally Reprogrammed Patient-derived Cells. Eur Urol. In press. https://doi.org/10.1016/j.eururo.2019.06.016. Eur. Urol. 2019. [Google Scholar] [CrossRef]
- Liu, X. Re: Kimmo Kettunen, Peter J. Bostrom, Tarja Lamminen; et al. Personalized Drug Sensitivity Screening for Bladder Cancer Using Conditionally Reprogrammed Patient-derived Cells. Eur Urol. In press. https://doi.org/10.1016/j.eururo.2019.06.016: Can Patient-derived Cancer Models Change the Costliest Cancer Type? Eur. Urol. 2019. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.; Li, M.; Wang, A.; Zhou, Y.; Li, P.; Wang, Y. Nanocarriers for TRAIL delivery: Driving TRAIL back on track for cancer therapy. Nanoscale 2017, 9, 13879–13904. [Google Scholar] [CrossRef] [PubMed]
- Vondálová Blanářová, O.; Šafaříková, B.; Herůdková, J.; Krkoška, M.; Tománková, S.; Kahounová, Z.; Anděra, L.; Bouchal, J.; Kharaishvili, G.; Král, M.; et al. Cisplatin or LA-12 enhance killing effects of TRAIL in prostate cancer cells through Bid-dependent stimulation of mitochondrial apoptotic pathway but not caspase-10. PLoS ONE 2017, 12, e0188584. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazi, A.M.; Kwon, S.Y.; Stanford, W.L. Stem cell sources for regenerative medicine. Methods Mol. Biol. 2009, 482, 55–90. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Foster, C.; Sayej, W.; Finck, C.M. Conditional Reprogramming of Pediatric Human Esophageal Epithelial Cells for Use in Tissue Engineering and Disease Investigation. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.J.I.; Hynds, R.E.; Gowers, K.H.C.; Tait, A.; Butler, C.R.; Hopper, C.; Burns, A.J.; Birchall, M.A.; Lowdell, M.; Janes, S.M. Using a Three-Dimensional Collagen Matrix to Deliver Respiratory Progenitor Cells to Decellularized Trachea In Vivo. Tissue Engineering. Part. Cmethods 2019, 25, 93–102. [Google Scholar] [CrossRef]
- LaRanger, R.; Peters-Hall, J.R.; Coquelin, M.; Alabi, B.R.; Chen, C.T.; Wright, W.E.; Shay, J.W. Reconstituting Mouse Lungs with Conditionally Reprogrammed Human Bronchial Epithelial Cells. Tissue Engineering. Part. A 2018, 24, 559–568. [Google Scholar] [CrossRef]
- Butler, C.R.; Hynds, R.E.; Gowers, K.H.; Lee Ddo, H.; Brown, J.M.; Crowley, C.; Teixeira, V.H.; Smith, C.M.; Urbani, L.; Hamilton, N.J.; et al. Rapid Expansion of Human Epithelial Stem Cells Suitable for Airway Tissue Engineering. Am. J. Respir. Crit. Care Med. 2016, 194, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Butler, C.R.; Hynds, R.E.; Gowers, K.H.C.; Brown, J.M.; Lee, D.D.H.; Teixeira, V.H.; Hamilton, N.J.; Birchall, M.A.; O’Callaghan, C.; Janes, S.M. Co-culture-expanded human basal epithelial stem cells for application in tracheal tissue engineering. Lancet 2016, 387, S23. [Google Scholar] [CrossRef]
- Gowers, K.H.C.; Hynds, R.E.; Thakrar, R.M.; Carroll, B.; Birchall, M.A.; Janes, S.M. Optimized isolation and expansion of human airway epithelial basal cells from endobronchial biopsy samples. J. Tissue Eng. Regen. Med. 2018, 12, e313–e317. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.W.; Rios, C.; Huang, C.; Wesolowska-Andersen, A.; Burchard, E.G.; O’Connor, B.P.; Fingerlin, T.E.; Nichols, D.; Reynolds, S.D.; Seibold, M.A. CRISPR-Cas9-mediated gene knockout in primary human airway epithelial cells reveals a proinflammatory role for MUC18. Gene Ther. 2015, 22, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Fenini, G.; Grossi, S.; Contassot, E.; Biedermann, T.; Reichmann, E.; French, L.E.; Beer, H.D. Genome Editing of Human Primary Keratinocytes by CRISPR/Cas9 Reveals an Essential Role of the NLRP1 Inflammasome in UVB Sensing. J. Investig. Dermatol. 2018, 138, 2644–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Li, C.H.; Chan, S.L.; Xu, F.; Feng, L.; Wang, Y.; Jiang, J.D.; Sung, J.J.; Cheng, C.H.; Chen, Y. A small-molecule modulator of the tumor-suppressor miR34a inhibits the growth of hepatocellular carcinoma. Cancer Res. 2014, 74, 6236–6247. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, P.; Gao, G.; Deng, J.; Pan, Z.; Wu, X.; Xie, G.; Yue, C.; Cho, C.H.; Ma, Y.; et al. Smac therapeutic Peptide nanoparticles inducing apoptosis of cancer cells for combination chemotherapy with Doxorubicin. Acs Appl. Mater. Interfaces 2015, 7, 8005–8012. [Google Scholar] [CrossRef]
- Li, M.; Li, L.; Zhang, L.; Hu, W.; Shen, J.; Xiao, Z.; Wu, X.; Chan, F.L.; Cho, C.H. 1,25-Dihydroxyvitamin D3 suppresses gastric cancer cell growth through VDR- and mutant p53-mediated induction of p21. Life Sci. 2017, 179, 88–97. [Google Scholar] [CrossRef]
- Li, X.; Vargas Buonfiglio, L.G.; Adam, R.J.; Stoltz, D.A.; Zabner, J.; Comellas, A.P. Cystic Fibrosis Transmembrane Conductance Regulator Potentiation as a Therapeutic Strategy for Pulmonary Edema: A Proof-of-Concept Study in Pigs. Crit. Care Med. 2017, 45, e1240–e1246. [Google Scholar] [CrossRef]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [Green Version]
- Gillet, J.P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef]
- Sugaya, M.; Takenoyama, M.; Osaki, T.; Yasuda, M.; Nagashima, A.; Sugio, K.; Yasumoto, K. Establishment of 15 cancer cell lines from patients with lung cancer and the potential tools for immunotherapy. Chest 2002, 122, 282–288. [Google Scholar] [CrossRef]
- Correa, B.R.S.; Hu, J.; Penalva, L.O.F.; Schlegel, R.; Rimm, D.L.; Galante, P.A.F.; Agarwal, S. Patient-derived conditionally reprogrammed cells maintain intra-tumor genetic heterogeneity. Sci. Rep. 2018, 8, 4097. [Google Scholar] [CrossRef] [PubMed]
- Dantas, A.N.; Morais, E.F.; Macedo, R.A.; Tinoco, J.M.; Morais Mde, L. Clinicopathological characteristics and perineural invasion in adenoid cystic carcinoma: A systematic review. Braz. J. Otorhinolaryngol. 2015, 81, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Ku, S.; Li, Q.; Azabdaftari, G.; Seliski, J.; Olson, B.; Netherby, C.S.; Tang, D.G.; Abrams, S.I.; Goodrich, D.W.; et al. Generation of a C57BL/6 MYC-Driven Mouse Model and Cell Line of Prostate Cancer. Prostate 2016, 76, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011, 242, 205–219. [Google Scholar] [CrossRef]
- Perez, G.F.; Rodriguez-Martinez, C.E.; Nino, G. Rhinovirus-Induced Airway Disease: A Model to Understand the Antiviral and Th2 Epithelial Immune Dysregulation in Childhood Asthma. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2015, 63, 792–795. [Google Scholar] [CrossRef]
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Generation of Human Nasal Epithelial Cell Spheroids for Individualized Cystic Fibrosis Transmembrane Conductance Regulator Study. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Wolf, S.; Perez, G.F.; Mukharesh, L.; Isaza, N.; Preciado, D.; Freishtat, R.J.; Pillai, D.; Rose, M.C.; Nino, G. Conditional reprogramming of pediatric airway epithelial cells: A new human model to investigate early-life respiratory disorders. Pediatric Allergy Immunol. Off. Publ. Eur. Soc. Pediatric Allergy Immunol. 2017, 28, 810–817. [Google Scholar] [CrossRef]
- Moorefield, E.C.; Blue, R.E.; Quinney, N.L.; Gentzsch, M.; Ding, S. Generation of renewable mouse intestinal epithelial cell monolayers and organoids for functional analyses. Bmc Cell Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Atala, A. Re: MYC Activation Cooperates with Vhl and Ink4a/Arf Loss to Induce Clear Cell Renal Cell Carcinoma. J. Urol. 2018, 199, 31. [Google Scholar] [CrossRef]
- Bailey, S.T.; Smith, A.M.; Kardos, J.; Wobker, S.E.; Wilson, H.L.; Krishnan, B.; Saito, R.; Lee, H.J.; Zhang, J.; Eaton, S.C.; et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat. Commun. 2017, 8, 15770. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Di Poto, C.; Kroemer, A.H.; Cui, W.; Roy, R.; Liu, X.; Ressom, H.W. Establishment of ornithine transcarbamylase deficiency-derived primary human hepatocyte with hepatic functions. Exp. Cell Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Di Poto, C.; Roy, R.; Liu, X.; Cui, W.; Kroemer, A.; Ressom, H.W. Highlight article: Long-term culture and characterization of patient-derived primary hepatocytes using conditional reprogramming. Exp. Biol. Med. (Maywood) 2019, 244, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Panaccione, A.; Chang, M.T.; Carbone, B.E.; Guo, Y.; Moskaluk, C.A.; Virk, R.K.; Chiriboga, L.; Prasad, M.L.; Judson, B.; Mehra, S.; et al. NOTCH1 and SOX10 are Essential for Proliferation and Radiation Resistance of Cancer Stem-Like Cells in Adenoid Cystic Carcinoma. Clin. Cancer Res. 2016, 22, 2083–2095. [Google Scholar] [CrossRef] [PubMed]
- Pollock, C.B.; McDonough, S.; Wang, V.S.; Lee, H.; Ringer, L.; Li, X.; Prandi, C.; Lee, R.J.; Feldman, A.S.; Koltai, H.; et al. Strigolactone analogues induce apoptosis through activation of p38 and the stress response pathway in cancer cell lines and in conditionally reprogrammed primary prostate cancer cells. Oncotarget 2014, 5, 1683–1698. [Google Scholar] [CrossRef]
- Kim, B.K.; Nam, S.W.; Min, B.S.; Ban, H.S.; Paik, S.; Lee, K.; Im, J.Y.; Lee, Y.; Park, J.T.; Kim, S.Y.; et al. Bcl-2-dependent synthetic lethal interaction of the IDF-11774 with the V0 subunit C of vacuolar ATPase (ATP6V0C) in colorectal cancer. Br. J. Cancer 2018, 119, 1347–1357. [Google Scholar] [CrossRef] [Green Version]
- Satthakarn, S.; Hladik, F.; Promsong, A.; Nittayananta, W. Vaginal innate immune mediators are modulated by a water extract of Houttuynia cordata Thunb. Bmc Complementary Altern. Med. 2015, 15, 183. [Google Scholar] [CrossRef]
- Alkhilaiwi, F.; Paul, S.; Zhou, D.; Zhang, X.; Wang, F.; Palechor-Ceron, N.; Wilson, K.; Guha, R.; Ferrer, M.; Grant, N.; et al. High-throughput screening identifies candidate drugs for the treatment of recurrent respiratory papillomatosis. Papillomavirus Res. 2019, 8, 100181. [Google Scholar] [CrossRef]
- Zhang, Z.; Bai, Q.; Chen, Y.; Ye, L.; Wu, X.; Long, X.; Ye, L.; Liu, J.; Li, H. Conditionally reprogrammed human normal bronchial epithelial cells express comparable levels of cytochromes p450 and are sensitive to BaP induction. Biochem. Biophys. Res. Commun. 2018, 503, 2132–2138. [Google Scholar] [CrossRef]
- Agarwal, S.; Hu, J.; Stanton, K.; Schalper, K.; Kluger, Y.; Zarrella, E.; Liu, X.; Schlegel, R.; Rimm, D.L. Abstract 1569: Next generation cell line models: Conditionally reprogrammed cells. Cancer Res. 2013, 73, 1569. [Google Scholar]
- Zhen-li, Y.; Ya-li, X.; Xiao-cui, B.; Hai-liang, F.; Yu-qin, L.; Qiang, S. Faciliated primary culture and amplification of breast cancer cells and their biological properties. Basic Clin. Med. 2017, V37, 224–229. [Google Scholar]
- Vaclova, T.; Maguire, S.; Pugh, M.; Barry, P.; Orr, N. Abstract 816: Molecular and genomic characterization of a newly established male breast cancer cell line. Tumor Biol. 2017, 77, 816. [Google Scholar]
- Kodack, D.P.; Farago, A.F.; Dastur, A.; Held, M.A.; Dardaei, L.; Friboulet, L.; von Flotow, F.; Damon, L.J.; Lee, D.; Parks, M.; et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep. 2017, 21, 3298–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.S.; Raffeld, M.; Moon, Y.W.; Xi, L.; Bianco, C.; Pham, T.; Lee, L.C.; Mitsudomi, T.; Yatabe, Y.; Okamoto, I.; et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J. Clin. Investig. 2014, 124, 3003–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Niederst, M.J.; Karlovich, C.A.; Wakelee, H.A.; Neal, J.W.; Mino-Kenudson, M.; Fulton, L.; Hata, A.N.; Lockerman, E.L.; Kalsy, A.; et al. Heterogeneity Underlies the Emergence of EGFRT790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third-Generation EGFR Inhibitor. Cancer Discov. 2015, 5, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Panaccione, A.; Zhang, Y.; Ryan, M.; Moskaluk, C.A.; Anderson, K.S.; Yarbrough, W.G.; Ivanov, S.V. MYB fusions and CD markers as tools for authentication and purification of cancer stem cells from salivary adenoid cystic carcinoma. Stem Cell Res. 2017, 21, 160–166. [Google Scholar] [CrossRef]
- Gentzsch, M.; Boyles, S.E.; Cheluvaraju, C.; Chaudhry, I.G.; Quinney, N.L.; Cho, C.; Dang, H.; Liu, X.; Schlegel, R.; Randell, S.H. Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2017, 56, 568–574. [Google Scholar] [CrossRef]
- Peters-Hall, J.R.; Coquelin, M.L.; Torres, M.J.; LaRanger, R.; Alabi, B.R.; Sho, S.; Calva-Moreno, J.F.; Thomas, P.J.; Shay, J.W. Long-term culture and cloning of primary human bronchial basal cells that maintain multipotent differentiation capacity and CFTR channel function. Am. J. Physiology. Lung Cell. Mol. Physiol. 2018, 315, l313–l327. [Google Scholar] [CrossRef]
- Martinovich, K.M.; Iosifidis, T.; Buckley, A.G.; Looi, K.; Ling, K.M.; Sutanto, E.N.; Kicic-Starcevich, E.; Garratt, L.W.; Shaw, N.C.; Montgomery, S.; et al. Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Sci. Rep. 2017, 7, 17971. [Google Scholar] [CrossRef]
- Reynolds, S.D.; Rios, C.; Wesolowska-Andersen, A.; Zhuang, Y.; Pinter, M.; Happoldt, C.; Hill, C.L.; Lallier, S.W.; Cosgrove, G.P.; Solomon, G.M.; et al. Airway Progenitor Clone Formation Is Enhanced by Y-27632–Dependent Changes in the Transcriptome. Am. J. Respir. Cell Mol. Biol. 2016, 55, 323–336. [Google Scholar] [CrossRef]
- Yu, F.; Lu, Y.; Tao, L.; Jiang, Y.Y.; Lin, D.C.; Wang, L.; Petersson, F.; Yoshiyama, H.; Koeffler, P.H.; Goh, B.C.; et al. Non-malignant epithelial cells preferentially proliferate from nasopharyngeal carcinoma biopsy cultured under conditionally reprogrammed conditions. Sci. Rep. 2017, 7, 17359. [Google Scholar] [CrossRef]
- Shay, J.W.; Peters-Hall, J.R.; Min, J.; Tedone, E.; Sho, S.; Siteni, S.; Mender, I. Human Lung Epithelial Cells Divide >200 Population Doublings without Engaging a Telomere Maintenance Mechanism. bioRxiv 2018. [Google Scholar] [CrossRef]
- Yang, M.; Wang, S.; Mengqian, L.I.; Liu, Z.; Liu, L.; Zhang, M.; Wang, F.J.O.B. The expansion of the oral mucosa epithelial cells in vitro in conditioned culture media supplemented with Y27632. Oral Biomed. 2015, 6, 90–94. [Google Scholar]
- Alamri, A.M.; Groeneveld, S.; Kang, K.; Dabydeen, S.; Wang, W.; Hennighausen, L.; Kallakury, B.; Liu, X.; Furth, P.A. Abstract 3918: Characterizing growth features, allograft generation and transcriptomes of cultured conditionally reprogrammed cells (CRC) prepared from primary triple negative cancer from Brca1-mutant mice. Cancer Res. 2014, 74, 3918. [Google Scholar] [CrossRef]
- Ligaba, S.B.; Khurana, A.; Graham, G.; Krawczyk, E.; Jablonski, S.; Petricoin, E.F.; Glazer, R.I.; Upadhyay, G. Multifactorial analysis of conditional reprogramming of human keratinocytes. PLoS ONE 2015, 10, e0116755. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bi, B.; Song, H.; Liu, L.; Zheng, H.; Wang, S.; Shen, Z. Proliferation of human hepatocellular carcinoma cells from surgically resected specimens under conditionally reprogrammed culture. Mol. Med. Rep. 2019, 19, 4623–4630. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conventional Cell Lines | Organoids | PDX | CRC | |
|---|---|---|---|---|
| Sample size | ||||
| FNA | − | +/− | − | +++ |
| Core Biopsy | − | + | − | +++ |
| Surgical Specimens | + | +++ | ++ | +++ |
| Timing | dozen days | 1-5 weeks | 1-5mont | 1-10 days |
| Success rate of initiation | + 0–10% | ++ (5–80%) | ++ (2–30%) | +++50–100% |
| Tumor type specific | ||||
| Rapid Expansion | +++ | ++ | + | +++ |
| Matched Normal con | − | + | - | + |
| Karyotypic stability | − | ++ | N/A | ++ |
| 3D growth | − | + | +++ | − |
| Representation of tumor | + | ++ | ++ | ++ |
| Genetic manipulation | +++ | ++ | − | ++ |
| Maintenance (passage) | +++ | ++ | + | +++ |
| LT drug screens | +++ | – | + | +++ |
| HT drug screens | +++ | ++ | − | +++ |
| Heterogeneity | − | ++ | +++ | ++ |
| Tumor–stroma interaction | − | − | ++ | − |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palechor-Ceron, N.; Krawczyk, E.; Dakic, A.; Simic, V.; Yuan, H.; Blancato, J.; Wang, W.; Hubbard, F.; Zheng, Y.-L.; Dan, H.; et al. Conditional Reprogramming for Patient-Derived Cancer Models and Next-Generation Living Biobanks. Cells 2019, 8, 1327. https://doi.org/10.3390/cells8111327
Palechor-Ceron N, Krawczyk E, Dakic A, Simic V, Yuan H, Blancato J, Wang W, Hubbard F, Zheng Y-L, Dan H, et al. Conditional Reprogramming for Patient-Derived Cancer Models and Next-Generation Living Biobanks. Cells. 2019; 8(11):1327. https://doi.org/10.3390/cells8111327
Chicago/Turabian StylePalechor-Ceron, Nancy, Ewa Krawczyk, Aleksandra Dakic, Vera Simic, Hang Yuan, Jan Blancato, Weisheng Wang, Fleesie Hubbard, Yun-Ling Zheng, Hancai Dan, and et al. 2019. "Conditional Reprogramming for Patient-Derived Cancer Models and Next-Generation Living Biobanks" Cells 8, no. 11: 1327. https://doi.org/10.3390/cells8111327
APA StylePalechor-Ceron, N., Krawczyk, E., Dakic, A., Simic, V., Yuan, H., Blancato, J., Wang, W., Hubbard, F., Zheng, Y. -L., Dan, H., Strome, S., Cullen, K., Davidson, B., Deeken, J. F., Choudhury, S., Ahn, P. H., Agarwal, S., Zhou, X., Schlegel, R., ... Liu, X. (2019). Conditional Reprogramming for Patient-Derived Cancer Models and Next-Generation Living Biobanks. Cells, 8(11), 1327. https://doi.org/10.3390/cells8111327