Isolation, Characterization, Differentiation and Immunomodulatory Capacity of Mesenchymal Stromal/Stem Cells from Human Perirenal Adipose Tissue

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Perirenal Adipose Tissue
2.2. Cell Isolation and Culture
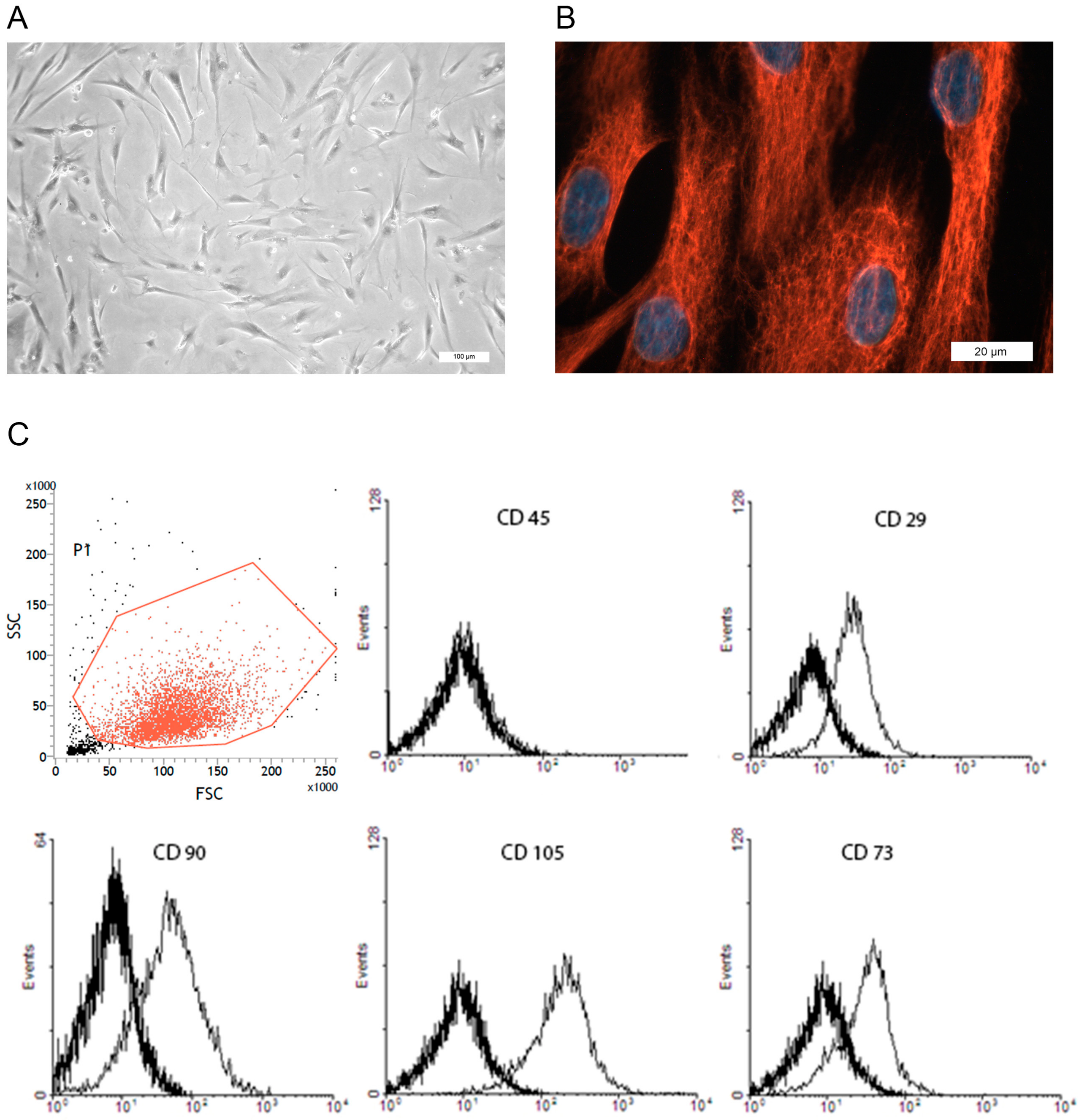
2.3. Cell Characterizations Using Flow Cytometry and Immunofluorescence Staining
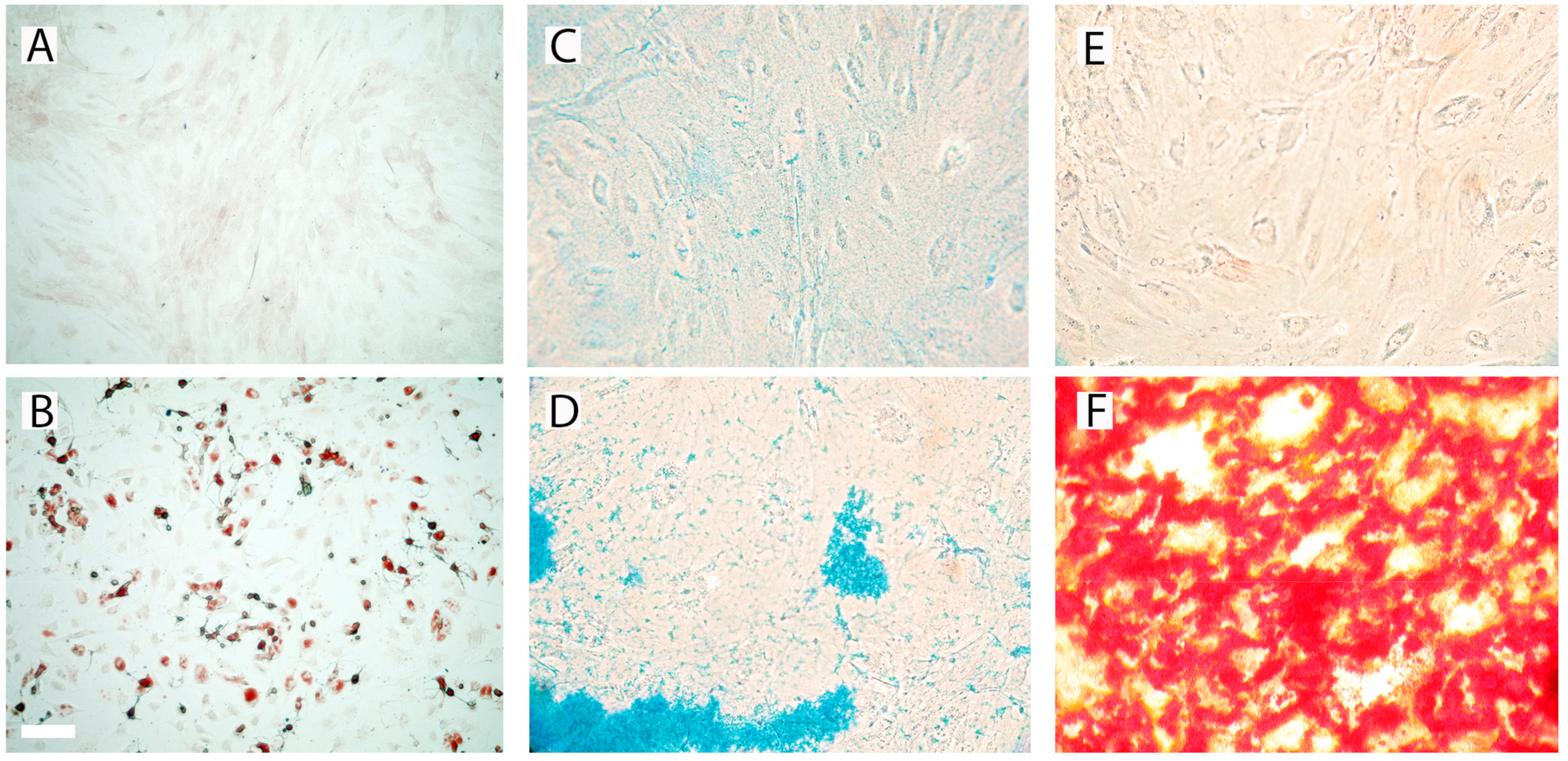
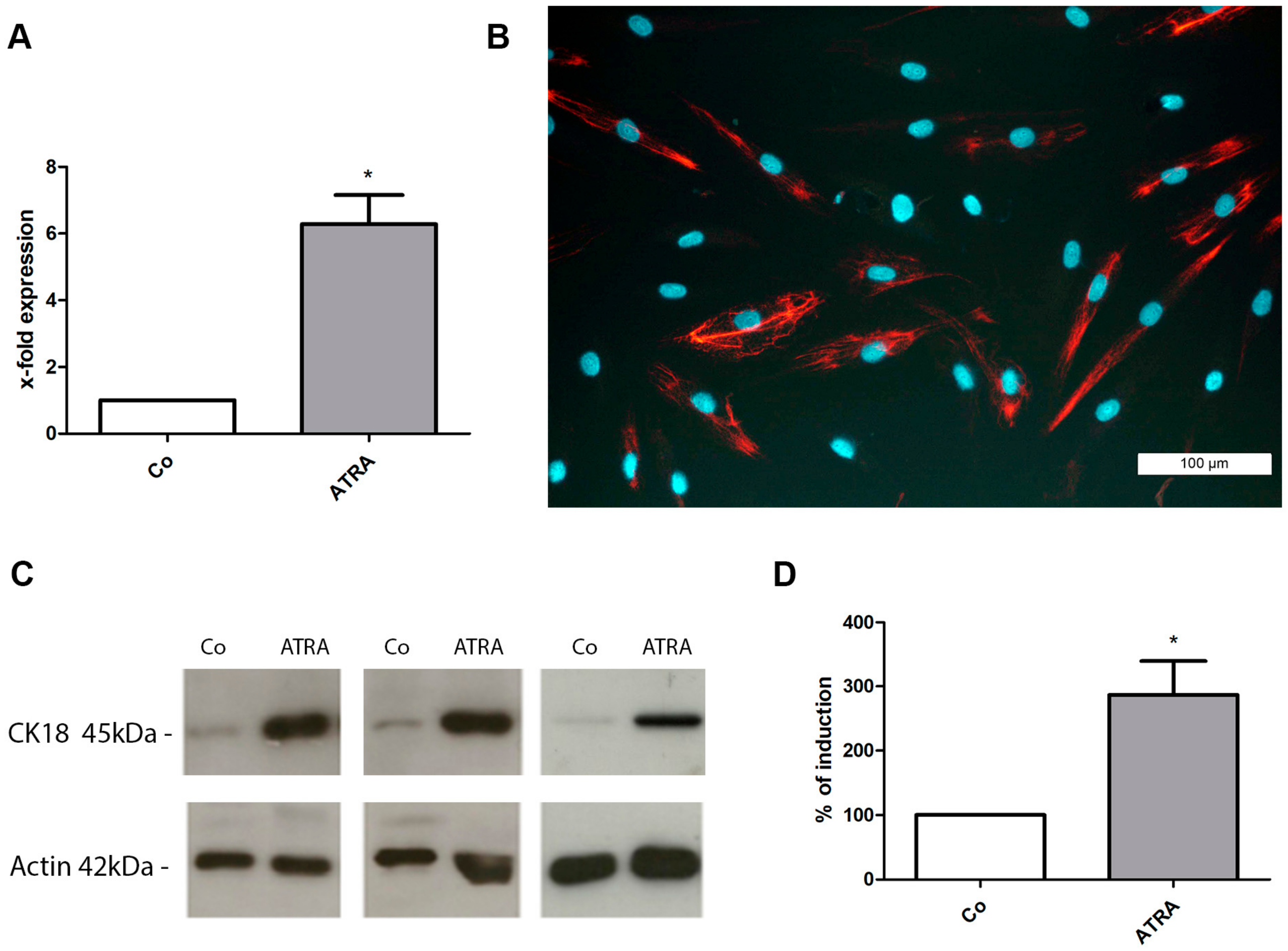
2.4. Induction of Cell Differentiation
2.5. Stimulation with LPS, LTA, and Cytokines
2.6. HCMV Infection
2.7. Cell Viability Assays
2.8. PCR
2.9. Western Blot
2.10. Immunoassay
2.11. Statistical Analysis
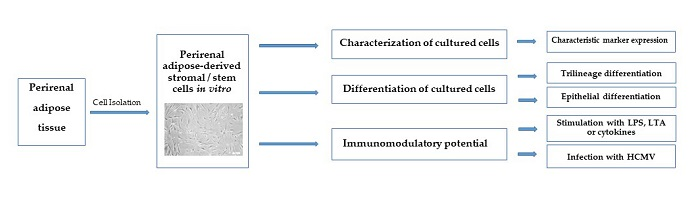
3. Results
3.1. Isolation and Characterization of prASCs
3.2. Differentiation of prASCs
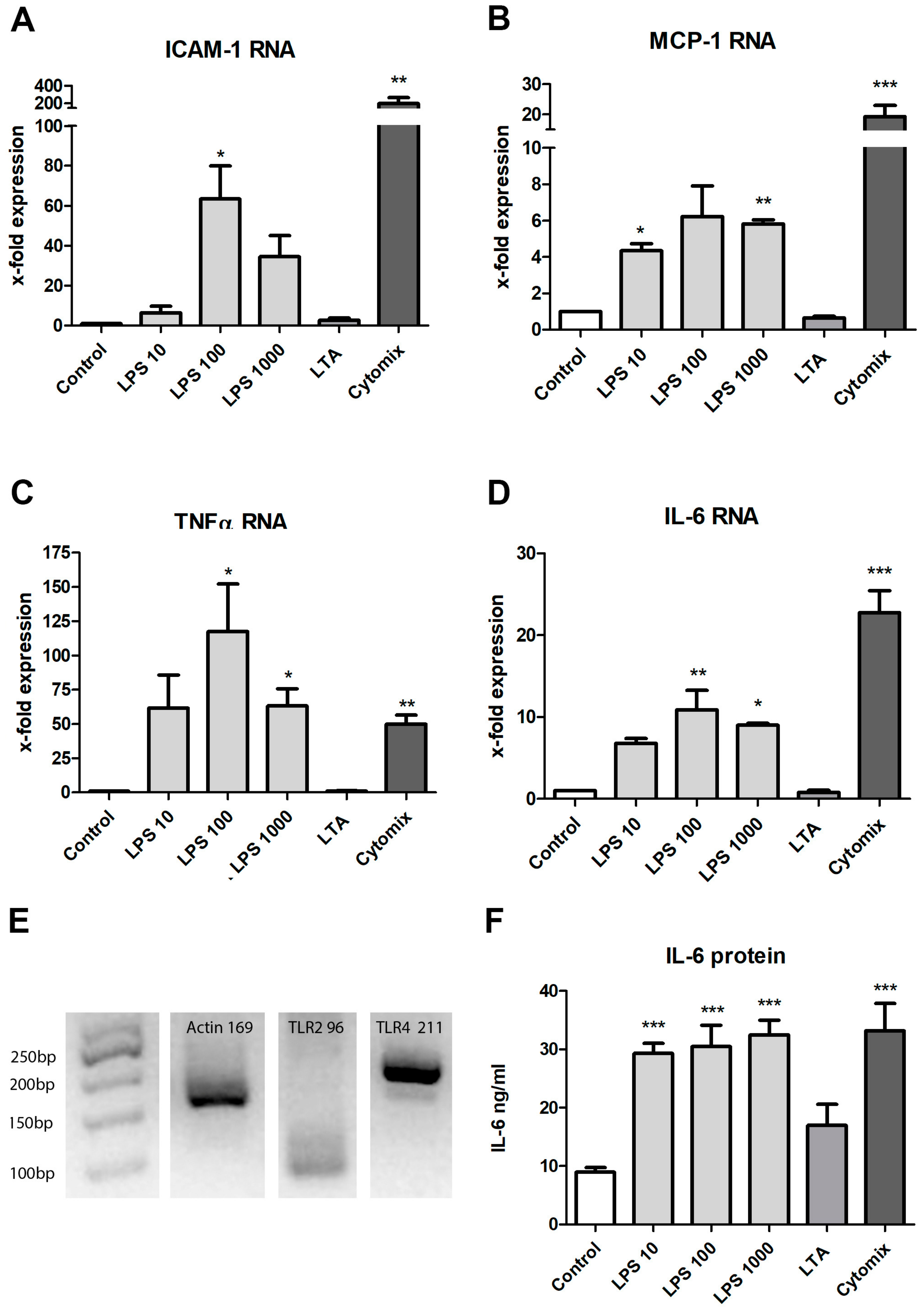
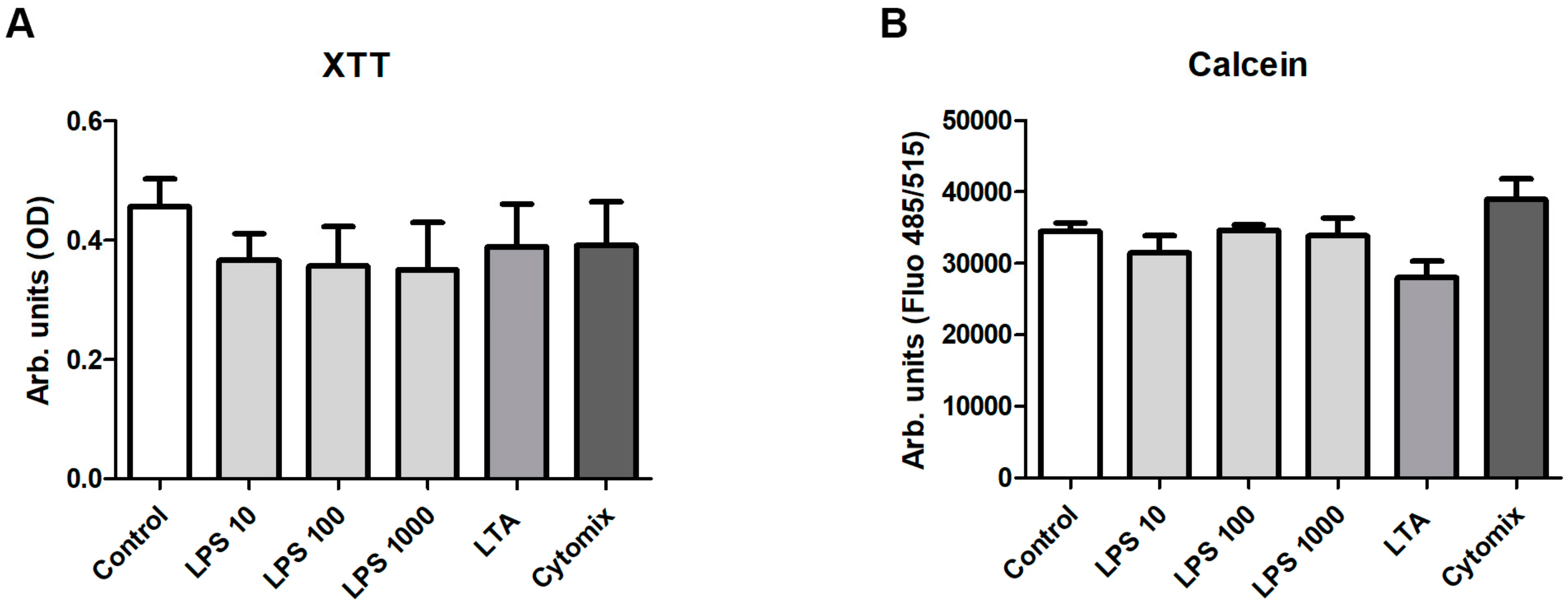
3.3. Stimulation with LPS, LTA and Cytokines
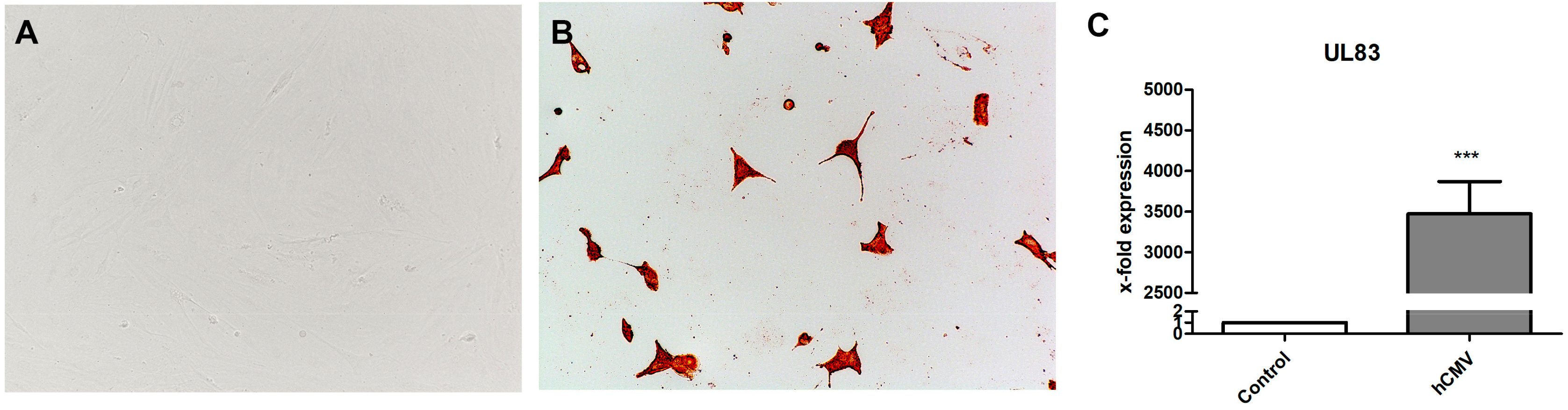
3.4. Infection with HCMV
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASCs | Adipose-derived MSCs |
| ATRA | All-trans retinoic acid |
| CK | Cytokeratin |
| CPE | Viral load dependent cytopathological effect |
| FBS | Fetal bovine serum |
| HCMV | Human cytomegalovirus |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IFNγ | γ−Interferon |
| IL | Interleukin |
| MCP-1 | Monocyte chemotactic protein |
| MSCs | Mesenchymal stromal/stem cells |
| MOI | Multiplicity of infection |
| PCR | Polymerase chain reaction |
| prASCs | ASCs from perirenal adipose tissue |
| qPCR | Quantitative real-time polymerase chain reaction |
| RT | Room temperature |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor-α |
| UL83 | HCMV-specific mRNA of UL83-coded phosphoprotein 65 |
References
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, P.C.; Geiger, H. Adipose-derived mesenchymal stromal/stem cells: Tissue localization, characterization, and heterogeneity. Stem Cells Int. 2012, 2012, 812693. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Russell, A.; Zubair, A.C. Effect of Comedications and Endotoxins on Mesenchymal Stem Cell Secretomes, Migratory and Immunomodulatory Capacity. J. Clin. Med. 2019, 8, 497. [Google Scholar] [CrossRef] [PubMed]
- Ponte, A.L.; Marais, E.; Gallay, N.; Langonné, A.; Delorme, B.; Hérault, O.; Charbord, P.; Domenech, J. The in vitro migration capacity of human bone marrow mesenchymal stem cells: Comparison of chemokine and growth factor chemotactic activities. Stem Cells 2007, 25, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef]
- Gao, F.; Chiu, S.M.; Motan, D.A.L.; Zhang, Z.; Chen, L.; Ji, H.-L.; Tse, H.-F.; Fu, Q.-L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef]
- Raicevic, G.; Rouas, R.; Najar, M.; Stordeur, P.; Boufker, H.I.; Bron, D.; Martiat, P.; Goldman, M.; Nevessignsky, M.T.; Lagneaux, L. Inflammation modifies the pattern and the function of Toll-like receptors expressed by human mesenchymal stromal cells. Hum. Immunol. 2010, 71, 235–244. [Google Scholar] [CrossRef]
- Liotta, F.; Angeli, R.; Cosmi, L.; Filì, L.; Manuelli, C.; Frosali, F.; Mazzinghi, B.; Maggi, L.; Pasini, A.; Lisi, V.; et al. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells 2008, 26, 279–289. [Google Scholar] [CrossRef]
- Cook, D.N.; Pisetsky, D.S.; Schwartz, D.A. Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 2004, 5, 975–979. [Google Scholar] [CrossRef]
- Lynes, M.D.; Tseng, Y.-H. Deciphering adipose tissue heterogeneity. Ann. N.Y. Acad. Sci. 2018, 1411, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.H.M.; Lam, K.S.L.; Xu, A. Heterogeneity of white adipose tissue: Molecular basis and clinical implications. Exp. Mol. Med. 2016, 48, e215. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-X.; Sun, W.; Kong, X.-Q. Perirenal Fat: A Unique Fat Pad and Potential Target for Cardiovascular Disease. Angiology 2019, 70, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.-Y.; Bandiera, R.; Serrels, A.; Martínez-Estrada, O.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell Biol. 2014, 16, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favre, G.; Grangeon-Chapon, C.; Raffaelli, C.; François-Chalmin, F.; Iannelli, A.; Esnault, V. Perirenal fat thickness measured with computed tomography is a reliable estimate of perirenal fat mass. PLoS ONE 2017, 12, e0175561. [Google Scholar] [CrossRef]
- Foster, M.C.; Hwang, S.-J.; Porter, S.A.; Massaro, J.M.; Hoffmann, U.; Fox, C.S. Fatty kidney, hypertension, and chronic kidney disease: The Framingham Heart Study. Hypertension 2011, 58, 784–790. [Google Scholar] [CrossRef]
- Czaja, K.; Kraeling, R.R.; Barb, C.R. Are hypothalamic neurons transsynaptically connected to porcine adipose tissue? Biochem. Biophys. Res. Commun. 2003, 311, 482–485. [Google Scholar] [CrossRef]
- Sundin, M.; Orvell, C.; Rasmusson, I.; Sundberg, B.; Ringdén, O.; Le Blanc, K. Mesenchymal stem cells are susceptible to human herpesviruses, but viral DNA cannot be detected in the healthy seropositive individual. Bone Marrow Transplant. 2006, 37, 1051–1059. [Google Scholar] [CrossRef]
- Smirnov, S.V.; Harbacheuski, R.; Lewis-Antes, A.; Zhu, H.; Rameshwar, P.; Kotenko, S.V. Bone-marrow-derived mesenchymal stem cells as a target for cytomegalovirus infection: Implications for hematopoiesis, self-renewal and differentiation potential. Virology 2007, 360, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Hoogduijn, M.J.; Crop, M.J.; Peeters, A.M.A.; van Osch, G.J.V.M.; Balk, A.H.M.M.; Ijzermans, J.N.M.; Weimar, W.; Baan, C.C. Human heart, spleen, and perirenal fat-derived mesenchymal stem cells have immunomodulatory capacities. Stem Cells Dev. 2007, 16, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Crop, M.J.; Baan, C.C.; Korevaar, S.S.; Ijzermans, J.N.M.; Alwayn, I.P.J.; Weimar, W.; Hoogduijn, M.J. Donor-derived mesenchymal stem cells suppress alloreactivity of kidney transplant patients. Transplantation 2009, 87, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Crop, M.J.; Baan, C.C.; Korevaar, S.S.; Ijzermans, J.N.M.; Weimar, W.; Hoogduijn, M.J. Human adipose tissue-derived mesenchymal stem cells induce explosive T-cell proliferation. Stem Cells Dev. 2010, 19, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Griesche, N.; Luttmann, W.; Luttmann, A.; Stammermann, T.; Geiger, H.; Baer, P.C. A simple modification of the separation method reduces heterogeneity of adipose-derived stem cells. Cells Tissues Organs (Print) 2010, 192, 106–115. [Google Scholar] [CrossRef]
- Baer, P.C.; Brzoska, M.; Geiger, H. Epithelial differentiation of human adipose-derived stem cells. Methods Mol. Biol. 2011, 702, 289–298. [Google Scholar] [CrossRef]
- Brzoska, M.; Geiger, H.; Gauer, S.; Baer, P. Epithelial differentiation of human adipose tissue-derived adult stem cells. Biochem. Biophys. Res. Commun. 2005, 330, 142–150. [Google Scholar] [CrossRef]
- Michaelis, M.; Paulus, C.; Löschmann, N.; Dauth, S.; Stange, E.; Doerr, H.W.; Nevels, M.; Cinatl, J. The multi-targeted kinase inhibitor sorafenib inhibits human cytomegalovirus replication. Cell. Mol. Life Sci. 2011, 68, 1079–1090. [Google Scholar] [CrossRef]
- Cinatl, J.; Weber, B.; Rabenau, H.; Gümbel, H.O.; Chenot, J.F.; Scholz, M.; Encke, A.; Doerr, H.W. In vitro inhibition of human cytomegalovirus replication in human foreskin fibroblasts and endothelial cells by ascorbic acid 2-phosphate. Antivir. Res. 1995, 27, 405–418. [Google Scholar] [CrossRef]
- Overath, J.M.; Gauer, S.; Obermüller, N.; Schubert, R.; Schäfer, R.; Geiger, H.; Baer, P.C. Short-term preconditioning enhances the therapeutic potential of adipose-derived stromal/stem cell-conditioned medium in cisplatin-induced acute kidney injury. Exp. Cell Res. 2016, 342, 175–183. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Baer, P.C.; Bereiter-Hahn, J.; Schubert, R.; Geiger, H. Differentiation status of human renal proximal and distal tubular epithelial cells in vitro: Differential expression of characteristic markers. Cells Tissues Organs (Print) 2006, 184, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Cleal, L.; Aldea, T.; Chau, Y.-Y. Fifty shades of white: Understanding heterogeneity in white adipose stem cells. Adipocyte 2017, 6, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Ritter, A.; Friemel, A.; Fornoff, F.; Adjan, M.; Solbach, C.; Yuan, J.; Louwen, F. Characterization of adipose-derived stem cells from subcutaneous and visceral adipose tissues and their function in breast cancer cells. Oncotarget 2015, 6, 34475–34493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, P.C.; Bereiter-Hahn, J.; Missler, C.; Brzoska, M.; Schubert, R.; Gauer, S.; Geiger, H. Conditioned medium from renal tubular epithelial cells initiates differentiation of human mesenchymal stem cells. Cell Prolif. 2009, 42, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xiong, L.; Wang, R.; Yuan, Q.; Xia, Y.; Sun, J.; Horch, R.E. In vitro expression of cytokeratin 18, 19 and tube formation of adipose-derived stem cells induced by the breast epithelial cell line HBL-100. J. Cell. Mol. Med. 2015, 19, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J.; Lombardo, E. Concise Review: Mesenchymal Stromal Cells Anno 2019: Dawn of the Therapeutic Era? Stem Cells Transl. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, R.; Spohn, G.; Baer, P.C. Mesenchymal Stem/Stromal Cells in Regenerative Medicine: Can Preconditioning Strategies Improve Therapeutic Efficacy? Transfus. Med. Hemother. 2016, 43, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Shin, T.-H.; Kim, H.-S. Current Strategies to Enhance Adipose Stem Cell Function: An Update. Int. J. Mol. Sci. 2019, 20, 3827. [Google Scholar] [CrossRef]
- Krampera, M.; Cosmi, L.; Angeli, R.; Pasini, A.; Liotta, F.; Andreini, A.; Santarlasci, V.; Mazzinghi, B.; Pizzolo, G.; Vinante, F.; et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006, 24, 386–398. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Heo, S.C.; Jeong, G.O.; Yoon, J.W.; Mo, W.M.; Lee, M.J.; Jang, I.-H.; Kwon, S.M.; Lee, J.S.; Kim, J.H. Tumor necrosis factor-α-activated mesenchymal stem cells promote endothelial progenitor cell homing and angiogenesis. Biochim. Biophys. Acta 2013, 1832, 2136–2144. [Google Scholar] [CrossRef]
- Crop, M.J.; Baan, C.C.; Korevaar, S.S.; Ijzermans, J.N.M.; Pescatori, M.; Stubbs, A.P.; van Ijcken, W.F.J.; Dahlke, M.H.; Eggenhofer, E.; Weimar, W.; et al. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin. Exp. Immunol. 2010, 162, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Nasef, A.; Zhang, Y.Z.; Mazurier, C.; Bouchet, S.; Bensidhoum, M.; Francois, S.; Gorin, N.C.; Lopez, M.; Thierry, D.; Fouillard, L.; et al. Selected Stro-1-enriched bone marrow stromal cells display a major suppressive effect on lymphocyte proliferation. Int. J. Lab. Hematol. 2009, 31, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Nasef, A.; Mathieu, N.; Chapel, A.; Frick, J.; François, S.; Mazurier, C.; Boutarfa, A.; Bouchet, S.; Gorin, N.-C.; Thierry, D.; et al. Immunosuppressive effects of mesenchymal stem cells: Involvement of HLA-G. Transplantation 2007, 84, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Jeong, H.J.; Lee, S.K.; Kim, S.-J. Lipopolysaccharide preconditioning of adipose-derived stem cells improves liver-regenerating activity of the secretome. Stem Cell Res. Ther. 2015, 6, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, T.; Salamon, A.; Adam, S.; Herzmann, N.; Taubenheim, J.; Peters, K. Impact of bacteria and bacterial components on osteogenic and adipogenic differentiation of adipose-derived mesenchymal stem cells. Exp. Cell Res. 2013, 319, 2883–2892. [Google Scholar] [CrossRef]
- Herzmann, N.; Salamon, A.; Fiedler, T.; Peters, K. Lipopolysaccharide induces proliferation and osteogenic differentiation of adipose-derived mesenchymal stromal cells in vitro via TLR4 activation. Exp. Cell Res. 2017, 350, 115–122. [Google Scholar] [CrossRef]
- Herzmann, N.; Salamon, A.; Fiedler, T.; Peters, K. Analysis of migration rate and chemotaxis of human adipose-derived mesenchymal stem cells in response to LPS and LTA in vitro. Exp. Cell Res. 2016, 342, 95–103. [Google Scholar] [CrossRef]
- Qiao, G.-H.; Zhao, F.; Cheng, S.; Luo, M.-H. Multipotent mesenchymal stromal cells are fully permissive for human cytomegalovirus infection. Virol. Sin. 2016, 31, 219–228. [Google Scholar] [CrossRef]
- Avanzi, S.; Leoni, V.; Rotola, A.; Alviano, F.; Solimando, L.; Lanzoni, G.; Bonsi, L.; Di Luca, D.; Marchionni, C.; Alvisi, G.; et al. Susceptibility of human placenta derived mesenchymal stromal/stem cells to human herpesviruses infection. PLoS ONE 2013, 8, e71412. [Google Scholar] [CrossRef]
- Zwezdaryk, K.J.; Ferris, M.B.; Strong, A.L.; Morris, C.A.; Bunnell, B.A.; Dhurandhar, N.V.; Gimble, J.M.; Sullivan, D.E. Human cytomegalovirus infection of human adipose-derived stromal/stem cells restricts differentiation along the adipogenic lineage. Adipocyte 2016, 5, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Soland, M.A.; Keyes, L.R.; Bayne, R.; Moon, J.; Porada, C.D.; St Jeor, S.; Almeida-Porada, G. Perivascular stromal cells as a potential reservoir of human cytomegalovirus. Am. J. Transplant. 2014, 14, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Baer, P.C.; Kuçi, S.; Krause, M.; Kuçi, Z.; Zielen, S.; Geiger, H.; Bader, P.; Schubert, R. Comprehensive phenotypic characterization of human adipose-derived stromal/stem cells and their subsets by a high throughput technology. Stem Cells Dev. 2013, 22, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Kosugi, I.; Meguro, S.; Iwashita, T. Pathogenesis of developmental anomalies of the central nervous system induced by congenital cytomegalovirus infection. Pathol. Int. 2017, 67, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Meisel, R.; Heseler, K.; Nau, J.; Schmidt, S.K.; Leineweber, M.; Pudelko, S.; Wenning, J.; Zimmermann, A.; Hengel, H.; Sinzger, C.; et al. Cytomegalovirus infection impairs immunosuppressive and antimicrobial effector functions of human multipotent mesenchymal stromal cells. Mediat. Inflamm. 2014, 2014, 898630. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Forward | Primer Reverse | Product Length (bp) | NCBI Reference Sequence |
|---|---|---|---|---|
| CK-18 | CAC AGT CTG CTG AGG TTG GA | CAA GCT GGC CTT CAG ATT TC | 110 | NM_000224 |
| ICAM-1 | CAGTGACTGTCACTCGAGATCT | CCTCTTGGCTTAGTCATGTGAC | 500 | NM_000201.3 |
| IL-6 | AAAGATGGCTGAAAAAGATGGATGC | ACAGCTCTGGCTTGTTCCTCACTAC | 150 | NM_000600.4 |
| MCP-1 | CCCCAGTCACCTGCTGTTAT | AGATCTCCTTGGCCACAATG | 135 | NM_002982.4 |
| TNFα | CGGGACGTGGAGCTGGCCGAGGAG | CACCAGCTGGTTATCTCTCAGCTC | 354 | NM_000594.4 |
| TLR-2 | GCCCATTGCTCTTTCACTGCTT | ATGACCCCCAAGACCCACAC | 96 | NM_003264.4 |
| TLR-4 | CCCGACAACCTCCCCTTCTC | GGGCTAAACTCTGGATGGGGT | 211 | NM_003266 |
| UL83 | GCAGCCACGGGATCGTACT | GGCTTTTACCTCACACGAGCATT | 159 | NC_006273 |
| β-actin | ACT GGA ACG GTG AAG GGT GAC | AGA GAA GTG GGG TGG CTT TT | 169 | NM_001101 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baer, P.C.; Koch, B.; Hickmann, E.; Schubert, R.; Cinatl, J., Jr.; Hauser, I.A.; Geiger, H. Isolation, Characterization, Differentiation and Immunomodulatory Capacity of Mesenchymal Stromal/Stem Cells from Human Perirenal Adipose Tissue. Cells 2019, 8, 1346. https://doi.org/10.3390/cells8111346
Baer PC, Koch B, Hickmann E, Schubert R, Cinatl J Jr., Hauser IA, Geiger H. Isolation, Characterization, Differentiation and Immunomodulatory Capacity of Mesenchymal Stromal/Stem Cells from Human Perirenal Adipose Tissue. Cells. 2019; 8(11):1346. https://doi.org/10.3390/cells8111346
Chicago/Turabian StyleBaer, Patrick C., Benjamin Koch, Elena Hickmann, Ralf Schubert, Jindrich Cinatl, Jr., Ingeborg A. Hauser, and Helmut Geiger. 2019. "Isolation, Characterization, Differentiation and Immunomodulatory Capacity of Mesenchymal Stromal/Stem Cells from Human Perirenal Adipose Tissue" Cells 8, no. 11: 1346. https://doi.org/10.3390/cells8111346
APA StyleBaer, P. C., Koch, B., Hickmann, E., Schubert, R., Cinatl, J., Jr., Hauser, I. A., & Geiger, H. (2019). Isolation, Characterization, Differentiation and Immunomodulatory Capacity of Mesenchymal Stromal/Stem Cells from Human Perirenal Adipose Tissue. Cells, 8(11), 1346. https://doi.org/10.3390/cells8111346