Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
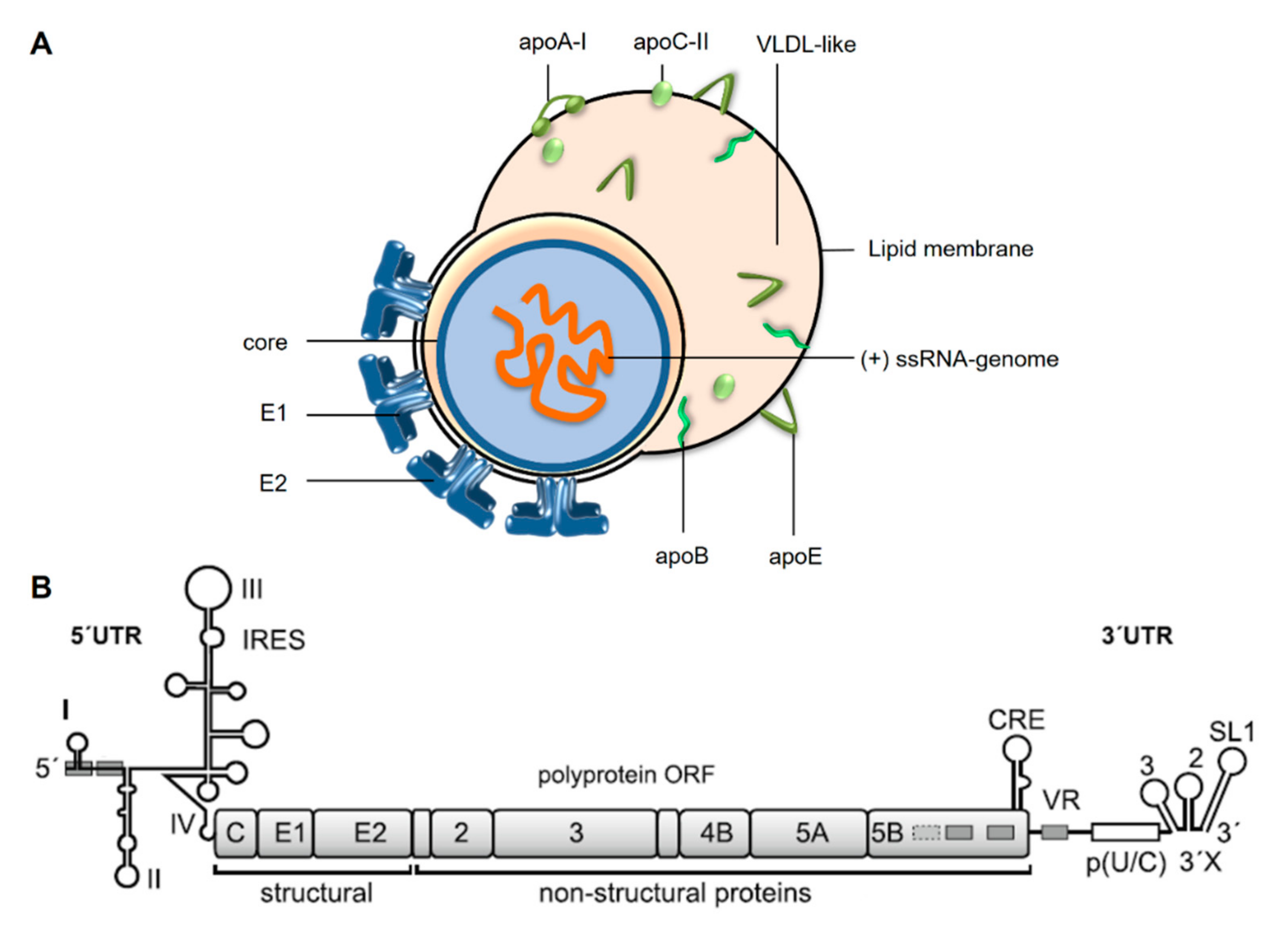
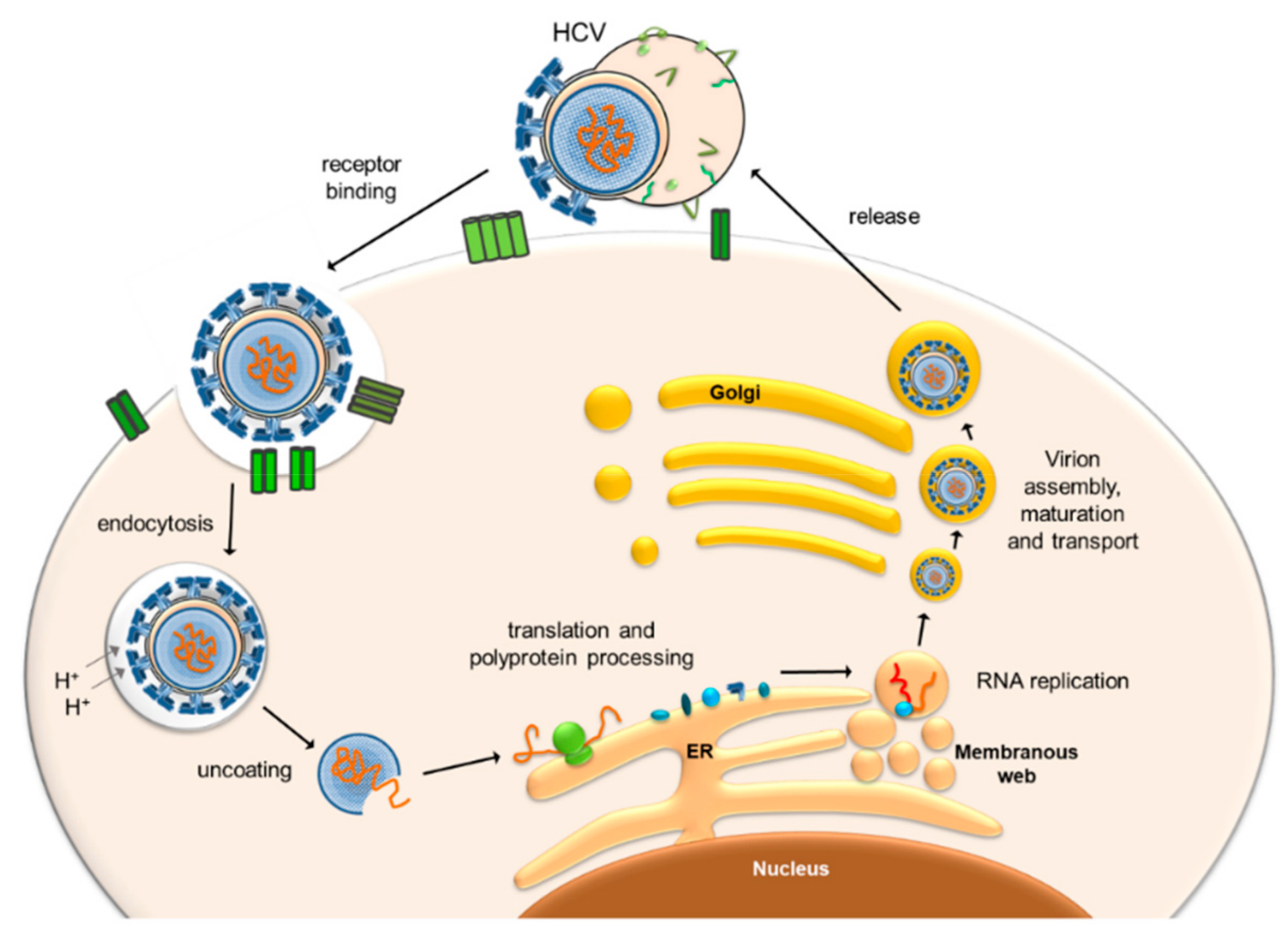
:1. Hepatitis C Virus Replication in the Liver

2. The Warburg Effect—Enhanced Glycolysis Flux to Provide Anabolic Metabolites in Cancer Cells
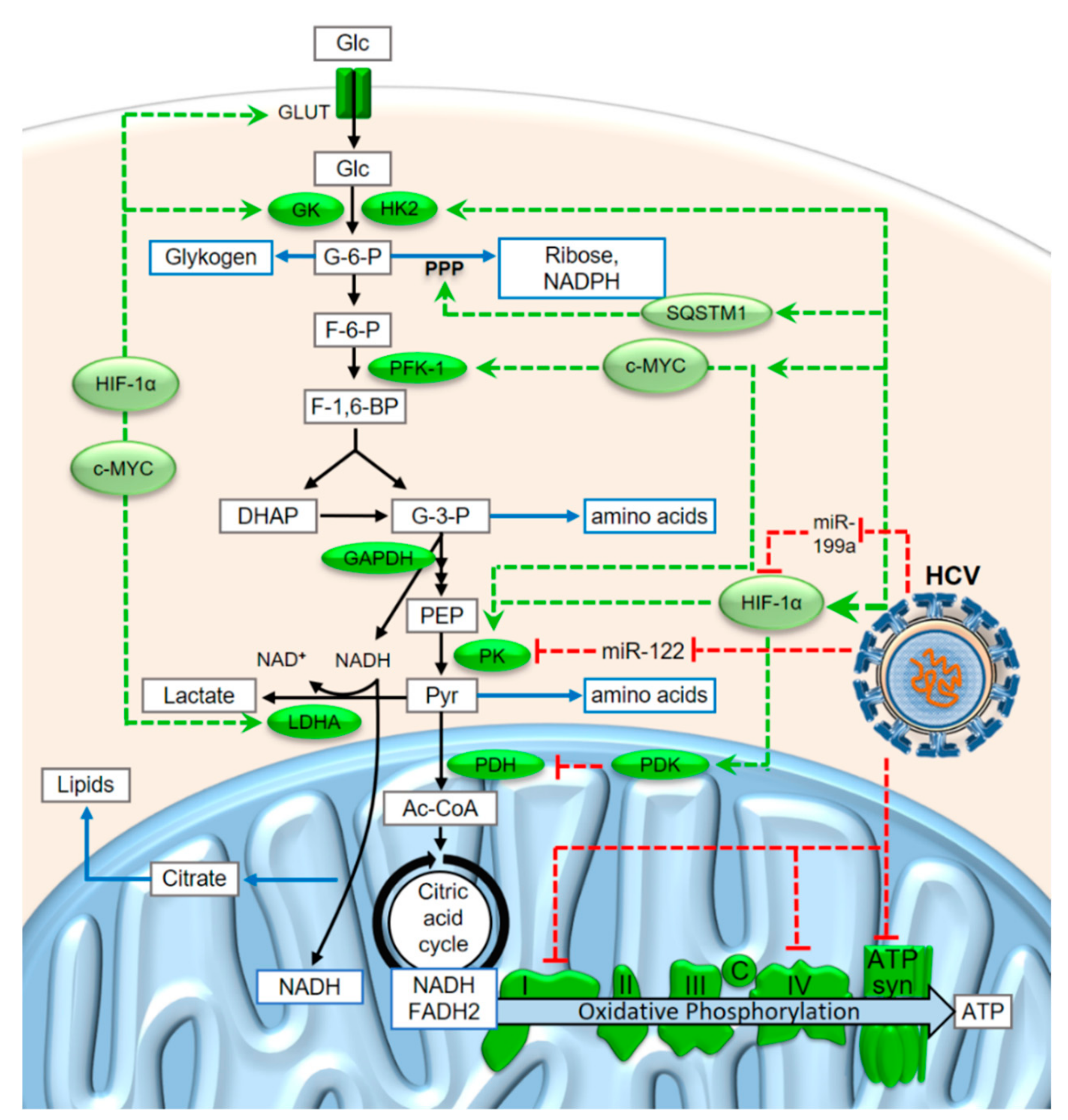
3. HCV Reprograms the Metabolism of Infected Cells towards a Cancer-Like State
4. A Revised View of the Metabolic Conditions in the Warburg Effect
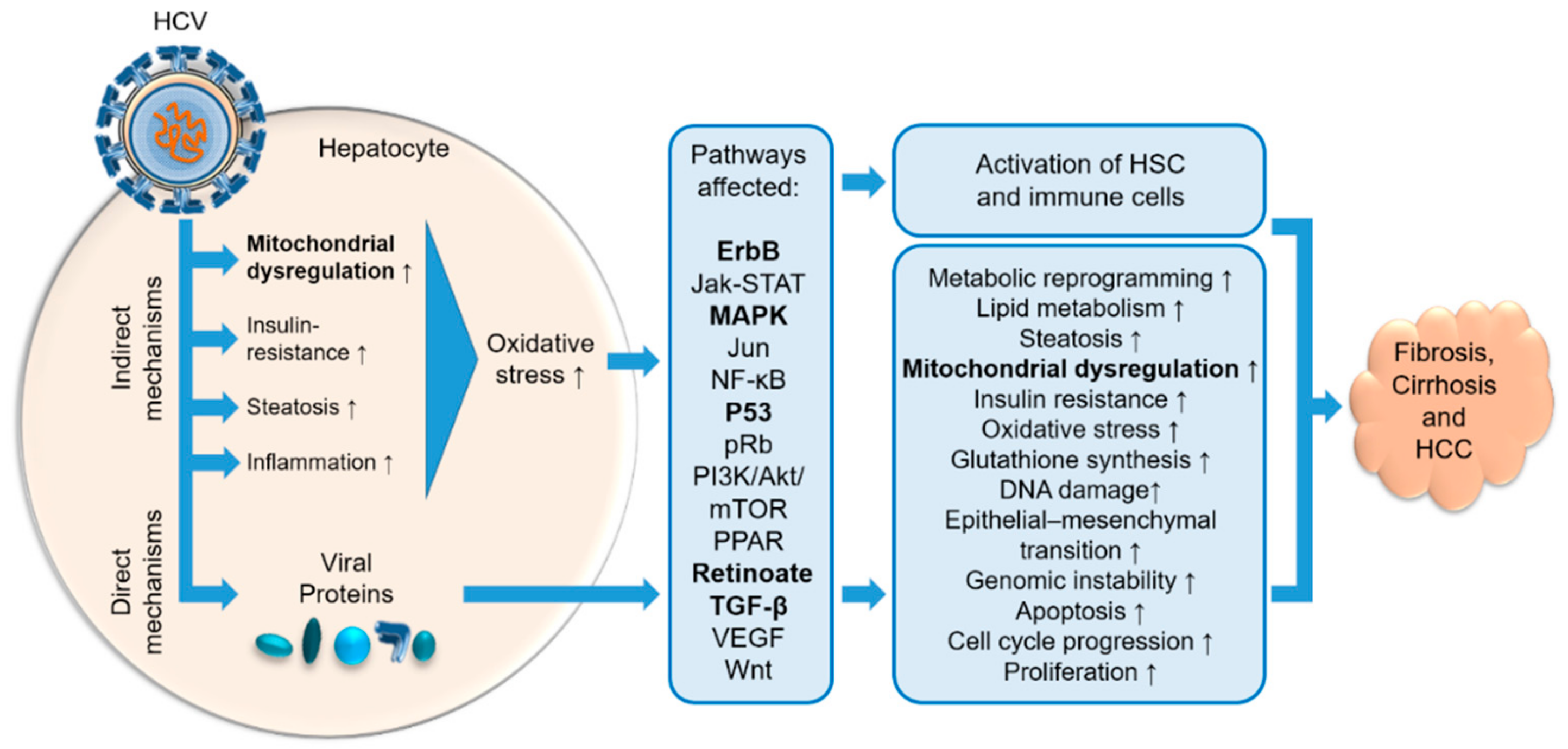
5. HCV Induces Fibrosis and Cancer Growth
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Virzi, A.; Roca Suarez, A.A.; Baumert, T.F.; Lupberger, J. Rewiring Host Signaling: Hepatitis C Virus in Liver Pathogenesis. Cold Spring Harb. Perspect. Med. 2019, 9, 37366. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Thimme, R. Insights from Antiviral Therapy into Immune Responses to Hepatitis B and C Virus Infection. Gastroenterology 2019, 156, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Waziry, R.; Hajarizadeh, B.; Grebely, J.; Amin, J.; Law, M.; Danta, M.; George, J.; Dore, G.J. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: A systematic review, meta-analyses, and meta-regression. J. Hepatol. 2017, 67, 1204–1212. [Google Scholar] [CrossRef]
- Guarino, M.; Vigano, L.; Ponziani, F.R.; Giannini, E.G.; Lai, Q.; Morisco, F. Special Interest Group on Hepatocellular, carcinoma and new anti-HCV therapies of the Italian Association for the Study of the Liver, Recurrence of hepatocellular carcinoma after direct acting antiviral treatment for hepatitis C virus infection: Literature review and risk analysis. Dig. Liver Dis. 2018, 50, 1105–1114. [Google Scholar]
- Hayes, C.N.; Zhang, P.; Zhang, Y.; Chayama, K. Molecular Mechanisms of Hepatocarcinogenesis Following Sustained Virological Response in Patients with Chronic Hepatitis C Virus Infection. Viruses 2018, 10, 531. [Google Scholar] [CrossRef]
- Na, S.K.; Song, B.C. Development and surveillance of hepatocellular carcinoma in patients with sustained virologic response after antiviral therapy for chronic hepatitis C. Clin. Mol. Hepatol. 2019, 25, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Vieyres, G.; Pietschmann, T. HCV Pit Stop at the Lipid Droplet: Refuel Lipids and Put on a Lipoprotein Coat before Exit. Cells 2019, 8, 233. [Google Scholar] [CrossRef]
- Gerold, G.; Moeller, R.; Pietschmann, T. Hepatitis C Virus Entry: Protein Interactions and Fusion Determinants Governing Productive Hepatocyte Invasion. Cold Spring Harb. Perspect. Med. 2019, 8, 36830. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C virus entry. Curr. Top Microbiol. Immunol. 2013, 369, 87–112. [Google Scholar]
- Miao, Z.; Xie, Z.; Miao, J.; Ran, J.; Feng, Y.; Xia, X. Regulated Entry of Hepatitis C Virus into Hepatocytes. Viruses 2017, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V. Hepatitis C Virus RNA Replication. Curr. Top Microbal. Immunol. 2013, 369, 167–198. [Google Scholar]
- Niepmann, M.; Shalamova, L.A.; Gerresheim, G.K.; Rossbach, O. Signals Involved in Regulation of Hepatitis C Virus RNA Genome Translation and Replication. Front. Microbiol. 2018, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Neufeldt, C.J.; Bartenschlager, R. Hepatitis C Virus Replication. Cold Spring Harb. Perspect. Med. 2019, 9, 37093. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar] [Green Version]
- Niepmann, M. Internal translation initiation of picornaviruses and hepatitis C virus. Biochim. Biophys. Acta 2009, 1789, 529–541. [Google Scholar] [CrossRef]
- Niepmann, M. Hepatitis C virus RNA translation. Curr. Top Microbiol. Immunol. 2013, 369, 143–166. [Google Scholar]
- Quade, N.; Boehringer, D.; Leibundgut, M.; van den Heuvel, J.; Ban, N. Cryo-EM structure of Hepatitis C virus IRES bound to the human ribosome at 3.9-A resolution. Nat. Commun. 2015, 6, 7646. [Google Scholar] [CrossRef]
- Garaigorta, U.; Chisari, F.V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009, 6, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Romero-Lopez, C.; Berzal-Herranz, A. The 5BSL3.2 Functional RNA Domain Connects Distant Regions in the Hepatitis C Virus Genome. Front. Microbiol. 2017, 8, 2093. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Strating, J.R.; van Kuppeveld, F.J. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 2017, 47, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Liu, Y.; Wimmer, E.; Paul, A.V. Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim. Biophys. Acta 2009, 1789, 495–517. [Google Scholar] [CrossRef]
- Mauger, D.M.; Golden, M.; Yamane, D.; Williford, S.; Lemon, S.M.; Martin, D.P.; Weeks, K.M. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. USA 2015, 112, 3692–3697. [Google Scholar] [CrossRef] [Green Version]
- Pirakitikulr, N.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. The Coding Region of the HCV Genome Contains a Network of Regulatory RNA Structures. Mol. Cell 2016, 62, 111–120. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Friebe, P.; Bartenschlager, R. Role of RNA structures in genome terminal sequences of the hepatitis C virus for replication and assembly. J. Virol. 2009, 83, 11989–11995. [Google Scholar] [CrossRef]
- Masante, C.; Mahias, K.; Lourenco, S.; Dumas, E.; Cahour, A.; Trimoulet, P.; Fleury, H.; Astier-Gin, T.; Ventura, M. Seven nucleotide changes characteristic of the hepatitis C virus genotype 3 5′ untranslated region: Correlation with reduced in vitro replication. J. Gen. Virol. 2008, 89, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. Embo. J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Fricke, M.; Dünnes, N.; Zayas, M.; Bartenschlager, R.; Niepmann, M.; Marz, M. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA 2015, 21, 1219–1232. [Google Scholar] [CrossRef] [Green Version]
- Jopling, C.L.; Schütz, S.; Sarnow, P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 2008, 4, 77–85. [Google Scholar] [CrossRef]
- Thibault, P.A.; Huys, A.; Amador-Canizares, Y.; Gailius, J.E.; Pinel, D.E.; Wilson, J.A. Regulation of Hepatitis C Virus Genome Replication by Xrn1 and MicroRNA-122 Binding to Individual Sites in the 5′ Untranslated Region. J. Virol. 2015, 89, 6294–6311. [Google Scholar] [CrossRef]
- Nieder-Röhrmann, A.; Dünnes, N.; Gerresheim, G.K.; Shalamova, L.A.; Herchenröther, A.; Niepmann, M. Cooperative enhancement of translation by two adjacent microRNA-122/Argonaute 2 complexes binding to the 5 untranslated region of Hepatitis C Virus RNA. J. Gen. Virol. 2017, 98, 212–224. [Google Scholar] [CrossRef]
- Roberts, A.P.; Lewis, A.P.; Jopling, C.L. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011, 39, 7716–7729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huys, A.; Thibault, P.A.; Wilson, J.A. Modulation of Hepatitis C Virus RNA Accumulation and Translation by DDX6 and miR-122 Are Mediated by Separate Mechanisms. PLoS ONE 2013, 8, e67437. [Google Scholar] [CrossRef] [PubMed]
- Schult, P.; Roth, H.; Adams, R.L.; Mas, C.; Imbert, L.; Orlik, C.; Ruggieri, A.; Pyle, A.M.; Lohmann, V. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 2018, 9, 2613. [Google Scholar] [CrossRef] [PubMed]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. USA 2011, 108, 3193–3198. [Google Scholar] [CrossRef] [PubMed]
- Nasheri, N.; Singaravelu, R.; Goodmurphy, M.; Lyn, R.K.; Pezacki, J.P. Competing roles of microRNA-122 recognition elements in hepatitis C virus RNA. Virology 2011, 410, 336–344. [Google Scholar] [CrossRef]
- Luna, J.M.; Scheel, T.K.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C Virus RNA Functionally Sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Gerresheim, G.K.; Dünnes, N.; Nieder-Röhrmann, A.; Shalamova, L.A.; Fricke, M.; Hofacker, I.; Höner Zu Siederdissen, C.; Marz, M.; Niepmann, M. microRNA-122 target sites in the hepatitis C virus RNA NS5B coding region and 3′ untranslated region: Function in replication and influence of RNA secondary structure. Cell Mol. Life Sci. 2017, 74, 747–760. [Google Scholar] [CrossRef]
- Lindenbach, B.D. Virion Assembly and Release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar] [Green Version]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef]
- Gerold, G.; Bruening, J.; Weigel, B.; Pietschmann, T. Protein Interactions during the Flavivirus and Hepacivirus Life Cycle. Mol. Cell. Proteom. 2017, 16, S75–S91. [Google Scholar] [CrossRef] [Green Version]
- Buenz, E.J.; Howe, C.L. Picornaviruses and cell death. Trends Microbiol. 2006, 14, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef] [PubMed]
- Dahari, H.; Ribeiro, R.M.; Rice, C.M.; Perelson, A.S. Mathematical modeling of subgenomic hepatitis C virus replication in Huh-7 cells. J. Virol. 2007, 81, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.E.; Dmitriev, S.E.; Terenin, I.M.; Prassolov, V.S.; Merrick, W.C.; Shatsky, I.N. Differential contribution of the m7G-cap to the 5′ end-dependent translation initiation of mammalian mRNAs. Nucleic Acids Res. 2009, 37, 6135–6147. [Google Scholar] [CrossRef]
- Sun, J.; Rajsbaum, R.; Yi, M. Immune and non-immune responses to hepatitis C virus infection. World J. Gastroenterol. 2015, 21, 10739–10748. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Wilhelm, J.; Gerresheim, G.K.; Shalamova, L.A.; Niepmann, M. Lnc-ITM2C-1 and GPR55 Are Proviral Host Factors for Hepatitis C Virus. Viruses 2019, 11, 549. [Google Scholar] [CrossRef]
- Yamane, D.; McGivern, D.R.; Masaki, T.; Lemon, S.M. Liver injury and disease pathogenesis in chronic hepatitis C. Curr. Top Microbiol. Immunol. 2013, 369, 263–288. [Google Scholar]
- Bandiera, S.; Billie Bian, C.; Hoshida, Y.; Baumert, T.F.; Zeisel, M.B. Chronic hepatitis C virus infection and pathogenesis of hepatocellular carcinoma. Curr. Opin. Virol. 2016, 20, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Bentaib, A.; de Tullio, P.; Chneiweiss, H.; Hermans, E.; Junier, M.P.; Leprince, P. Metabolic reprogramming in transformed mouse cortical astrocytes: A proteomic study. J. Proteom. 2015, 113, 292–314. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, N.; Kalliampakou, K.I.; Kotta-Loizou, I.; Befani, C.; Liakos, P.; Simos, G.; Mentis, A.F.; Kalliaropoulos, A.; Doumba, P.P.; Smirlis, D.; et al. Low oxygen tension enhances hepatitis C virus replication. J. Virol. 2013, 87, 2935–2948. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.Z.; Qu, S.B.; Wang, D.S. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J. Gastroenterol. 2016, 22, 9933–9943. [Google Scholar] [CrossRef]
- Marbaniang, C.; Kma, L. Dysregulation of Glucose Metabolism by Oncogenes and Tumor Suppressors in Cancer Cells. Asian Pac. J. Cancer Prev. 2018, 19, 2377–2390. [Google Scholar]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. ‘Why do tumour cells glycolyse?’ From glycolysis through citrate to lipogenesis. Mol. Cell. Biochem. 2005, 280, 1–8. [Google Scholar] [CrossRef]
- Xie, J.; Dai, C.; Hu, X. Evidence That Does Not Support Pyruvate Kinase M2 (PKM2)-catalyzed Reaction as a Rate-limiting Step in Cancer Cell Glycolysis. J. Biol. Chem. 2016, 291, 8987–8999. [Google Scholar] [CrossRef] [Green Version]
- San-Millan, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J.J.; Stryer, L. Biochemistry, 9th ed.; WH Freeman: New York, NY, USA, 2019. [Google Scholar]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef]
- Wong, M.C.; Jiang, J.Y.; Goggins, W.B.; Liang, M.; Fang, Y.; Fung, F.D.; Leung, C.; Wang, H.H.; Wong, G.L.; Wong, V.W.; et al. International incidence and mortality trends of liver cancer: A global profile. Sci. Rep. 2017, 7, 45846. [Google Scholar] [CrossRef] [PubMed]
- Irshad, M.; Gupta, P.; Irshad, K. Molecular basis of hepatocellular carcinoma induced by hepatitis C virus infection. World J. Hepatol. 2017, 9, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Gerresheim, G.K.; Bathke, J.; Michel, A.M.; Andreev, D.E.; Shalamova, L.A.; Rossbach, O.; Hu, P.; Glebe, D.; Fricke, M.; Marz, M.; et al. Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. Int. J. Mol. Sci. 2019, 20, 1321. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Fricke, M.; Gerst, R.; Ibrahim, B.; Niepmann, M.; Marz, M. Global importance of RNA secondary structures in protein coding sequences. Bioinformatics 2019, 35, 579–583. [Google Scholar] [CrossRef]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [Green Version]
- Wolski, D.; Foote, P.K.; Chen, D.Y.; Lewis-Ximenez, L.L.; Fauvelle, C.; Aneja, J.; Walker, A.; Tonnerre, P.; Torres-Cornejo, A.; Kvistad, D.; et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8(+) T Cells in Chronic versus Acute Infection. Immunity 2017, 47, 648–663. [Google Scholar] [CrossRef]
- Wolski, D.; Lauer, G.M. Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection. Viruses 2019, 11, 683. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.S.; Jeon, J.H.; Choi, Y.K.; Jang, S.Y.; Park, S.Y.; Kim, S.W.; Byun, J.K.; Kim, M.K.; Lee, S.; Shin, E.C.; et al. Pyruvate dehydrogenase kinase regulates hepatitis C virus replication. Sci. Rep. 2016, 6, 30846. [Google Scholar] [CrossRef]
- Bishop, T.; Ratcliffe, P.J. Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: A historical overview and future perspectives. Hypoxia 2014, 2, 197–213. [Google Scholar]
- Kaelin, W.G., Jr. The VHL Tumor Suppressor Gene: Insights into Oxygen Sensing and Cancer. Trans Am. Clin. Clim. Assoc. 2017, 128, 298–307. [Google Scholar]
- Meoni, G.; Lorini, S.; Monti, M.; Madia, F.; Corti, G.; Luchinat, C.; Zignego, A.L.; Tenori, L.; Gragnani, L. The metabolic fingerprints of HCV and HBV infections studied by Nuclear Magnetic Resonance Spectroscopy. Sci. Rep. 2019, 9, 4128. [Google Scholar] [CrossRef]
- Ramiere, C.; Rodriguez, J.; Enache, L.S.; Lotteau, V.; Andre, P.; Diaz, O. Activity of hexokinase is increased by its interaction with hepatitis C virus protein NS5A. J. Virol. 2014, 88, 3246–3254. [Google Scholar] [CrossRef]
- Mannova, P.; Beretta, L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell survival and viral replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef]
- Awad, A.; Gassama-Diagne, A. PI3K/SHIP2/PTEN pathway in cell polarity and hepatitis C virus pathogenesis. World J. Hepatol. 2017, 9, 18–29. [Google Scholar] [CrossRef]
- Peng, L.; Liang, D.; Tong, W.; Li, J.; Yuan, Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J. Biol. Chem. 2010, 285, 20870–20881. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Harak, C.; Benkheil, M.; Khan, H.; Leyssen, P.; Andrews, M.; Lohmann, V.; Neyts, J. PI4KIII inhibitor enviroxime impedes the replication of the hepatitis C virus by inhibiting PI3 kinases. J. Antimicrob. Chemother. 2018, 73, 3375–3384. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Casanova, C.; Fischer, B.; Weiss, A.; Fofana, I.; Fontaine, N.; Fujiwara, T.; Renaud, M.; Kopp, A.; Schuster, C.; et al. PI4K-beta and MKNK1 are regulators of hepatitis C virus IRES-dependent translation. Sci. Rep. 2015, 5, 13344. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hoffman, B.; Liu, Q. PI3K-Akt signaling pathway upregulates hepatitis C virus RNA translation through the activation of SREBPs. Virology 2016, 490, 99–108. [Google Scholar] [CrossRef]
- Murakami, Y.; Aly, H.H.; Tajima, A.; Inoue, I.; Shimotohno, K. Regulation of the hepatitis C virus genome replication by miR-199a*. J. Hepatol. 2009, 50, 453–460. [Google Scholar] [CrossRef]
- Pietschmann, T. Regulation of hepatitis C virus replication by microRNAs. J. Hepatol. 2009, 50, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.M.; Xu, Z.; Shek, F.H.; Wong, K.F.; Lee, N.P.; Poon, R.T.; Chen, J.; Luk, J.M. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS ONE 2014, 9, e86872. [Google Scholar] [CrossRef]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef]
- Sugiyama, K.; Ebinuma, H.; Nakamoto, N.; Sakasegawa, N.; Murakami, Y.; Chu, P.S.; Usui, S.; Ishibashi, Y.; Wakayama, Y.; Taniki, N.; et al. Prominent steatosis with hypermetabolism of the cell line permissive for years of infection with hepatitis C virus. PLoS ONE 2014, 9, e94460. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchéz-Martín, P.; Saito, T.; Komatsu, M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Hwang, L.Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.M.; Govindarajan, S.; Arakawa, K.; Yu, M.C. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer 2004, 101, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Ray, R. Hepatitis C virus infection and insulin resistance. World J. Diabetes 2014, 5, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.L. Metabolic alterations and hepatitis C: From bench to bedside. World J. Gastroenterol. 2016, 22, 1461–1476. [Google Scholar] [CrossRef] [PubMed]
- Lerat, H.; Imache, M.R.; Polyte, J.; Gaudin, A.; Mercey, M.; Donati, F.; Baudesson, C.; Higgs, M.R.; Picard, A.; Magnan, C.; et al. Hepatitis C virus induces a prediabetic state by directly impairing hepatic glucose metabolism in mice. J. Biol. Chem. 2017, 292, 12860–12873. [Google Scholar] [CrossRef] [Green Version]
- Shoji, I.; Deng, L.; Hotta, H. Molecular mechanism of hepatitis C virus-induced glucose metabolic disorders. Front. Microbiol. 2011, 2, 278. [Google Scholar] [CrossRef]
- Chen, J.; Wang, N.; Dong, M.; Guo, M.; Zhao, Y.; Zhuo, Z.; Zhang, C.; Chi, X.; Pan, Y.; Jiang, J.; et al. The Metabolic Regulator Histone Deacetylase 9 Contributes to Glucose Homeostasis Abnormality Induced by Hepatitis C Virus Infection. Diabetes 2015, 64, 4088–4098. [Google Scholar] [CrossRef] [Green Version]
- Mukhtar, N.A.; Bacchetti, P.; Ayala, C.E.; Melgar, J.; Christensen, S.; Maher, J.J.; Khalili, M. Insulin sensitivity and variability in hepatitis C virus infection using direct measurement. Dig. Dis. Sci. 2013, 58, 1141–1148. [Google Scholar] [CrossRef]
- Zhou, Z.H.; McCarthy, D.B.; O’Connor, C.M.; Reed, L.J.; Stoops, J.K. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. USA 2001, 98, 14802–14807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, F.; Golbik, R.; Brauer, J.; Lilie, H.; Schroder-Tittmann, K.; Hinze, E.; Korotchkina, L.G.; Patel, M.S.; Tittmann, K. Direct kinetic evidence for half-of-the-sites reactivity in the E1 component of the human pyruvate dehydrogenase multienzyme complex through alternating sites cofactor activation. Biochemistry 2006, 45, 12775–12785. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr.; Jenkins, W.T. Alanine aminotransferase. I. Purification and properties. J. Biol. Chem. 1967, 242, 91–100. [Google Scholar]
- Bergmeyer, H.U.; Scheibe, P.; Wahlefeld, A.W. Optimization of methods for aspartate aminotransferase and alanine aminotransferase. Clin. Chem. 1978, 24, 58–73. [Google Scholar]
- Fernandez-Velasco, D.A.; Garza-Ramos, G.; Ramirez, L.; Shoshani, L.; Darszon, A.; Tuena de Gomez-Puyou, M.; Gomez-Puyou, A. Activity of heart and muscle lactate dehydrogenases in all-aqueous systems and in organic solvents with low amounts of water. Effect of guanidine chloride. Eur. J. Biochem. 1992, 205, 501–508. [Google Scholar] [CrossRef]
- Crow, K.E.; Braggins, T.J.; Hardman, M.J. Human liver cytosolic malate dehydrogenase: Purification, kinetic properties, and role in ethanol metabolism. Arch. Biochem. Biophys. 1983, 225, 621–629. [Google Scholar] [CrossRef]
- Bartosch, B.; Thimme, R.; Blum, H.E.; Zoulim, F. Hepatitis C virus-induced hepatocarcinogenesis. J. Hepatol. 2009, 51, 810–820. [Google Scholar] [CrossRef] [Green Version]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.W.; Jang, J.Y.; Chung, R.T. Hepatitis C virus and hepatocarcinogenesis. Clin. Mol. Hepatol. 2012, 18, 347–356. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Quarato, G.; Ripoli, M.; D’Aprile, A.; Scrima, R.; Cela, O.; Boffoli, D.; Moradpour, D.; Capitanio, N. HCV infection induces mitochondrial bioenergetic unbalance: Causes and effects. Biochim. Biophys. Acta 2009, 1787, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [Green Version]
- Bedimo, R.; Maalouf, N.M.; Lo Re, V., 3rd. Hepatitis C virus coinfection as a risk factor for osteoporosis and fracture. Curr. Opin. Hiv Aids 2016, 11, 285–293. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Thongprayoon, C.; Panjawatanan, P.; Phatharacharukul, P.; Ungprasert, P. Hepatitis C virus infection and risk of osteoporosis: A meta-analysis. Saudi, J. Gastroenterol. 2017, 23, 216–221. [Google Scholar] [CrossRef]
- Lin, M.S.; Chen, P.H.; Wang, P.C.; Lin, H.S.; Huang, T.J.; Chang, S.T.; Chiu, W.N.; Chen, M.Y. Association between hepatitis C virus infection and osteoporotic fracture risk among postmenopausal women: A cross-sectional investigation in Taiwan. BMJ Open 2019, 9, e021990. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, J.; Huang, M.; Xu, G.; Wei, W.; Qin, H. Inhibition of miR-148a-3p resists hepatocellular carcinoma progress of hepatitis C virus infection through suppressing c-Jun and MAPK pathway. J. Cell. Mol. Med. 2019, 23, 1415–1426. [Google Scholar] [CrossRef]
- Roderfeld, M.; Padem, S.; Lichtenberger, J.; Quack, T.; Weiskirchen, R.; Longerich, T.; Schramm, G.; Churin, Y.; Irungbam, K.; Tschuschner, A.; et al. Schistosoma mansoni Egg-Secreted Antigens Activate Hepatocellular Carcinoma-Associated Transcription Factors c-Jun and STAT3 in Hamster and Human Hepatocytes. Hepatology 2018, 7, 30192. [Google Scholar] [CrossRef]
- Bosch, F.X.; Ribes, J.; Diaz, M.; Cleries, R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004, 127, 5–16. [Google Scholar] [CrossRef]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.J.; Wang, L.; Ren, H.; Cao, J.; Li, L.; Ke, J.S.; Qi, Z.T. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Exp. Cell Res. 2005, 305, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Ghozlan, H.; Abdel-Kader, O. Activation of RB/E2F signaling pathway is required for the modulation of hepatitis C virus core protein-induced cell growth in liver and non-liver cells. Cell Signal. 2004, 16, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Syed, V. TGF-beta Signaling in Cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Schulze-Krebs, A.; Preimel, D.; Popov, Y.; Bartenschlager, R.; Lohmann, V.; Pinzani, M.; Schuppan, D. Hepatitis C virus-replicating hepatocytes induce fibrogenic activation of hepatic stellate cells. Gastroenterology 2005, 129, 246–258. [Google Scholar] [CrossRef]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.C.; Zolfaghari, R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 2011, 31, 65–87. [Google Scholar] [CrossRef]
- Yanagitani, A.; Yamada, S.; Yasui, S.; Shimomura, T.; Murai, R.; Murawaki, Y.; Hashiguchi, K.; Kanbe, T.; Saeki, T.; Ichiba, M.; et al. Retinoic acid receptor alpha dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004, 40, 366–375. [Google Scholar] [CrossRef]
- Wang, L.; Potter, J.J.; Rennie-Tankersley, L.; Novitskiy, G.; Sipes, J.; Mezey, E. Effects of retinoic acid on the development of liver fibrosis produced by carbon tetrachloride in mice. Biochim. Biophys. Acta 2007, 1772, 66–71. [Google Scholar] [CrossRef] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerresheim, G.K.; Roeb, E.; Michel, A.M.; Niepmann, M. Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells. Cells 2019, 8, 1410. https://doi.org/10.3390/cells8111410
Gerresheim GK, Roeb E, Michel AM, Niepmann M. Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells. Cells. 2019; 8(11):1410. https://doi.org/10.3390/cells8111410
Chicago/Turabian StyleGerresheim, Gesche K., Elke Roeb, Audrey M. Michel, and Michael Niepmann. 2019. "Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells" Cells 8, no. 11: 1410. https://doi.org/10.3390/cells8111410
APA StyleGerresheim, G. K., Roeb, E., Michel, A. M., & Niepmann, M. (2019). Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells. Cells, 8(11), 1410. https://doi.org/10.3390/cells8111410