Augmenter of Liver Regeneration Reduces Ischemia Reperfusion Injury by Less Chemokine Expression, Gr-1 Infiltration and Oxidative Stress

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
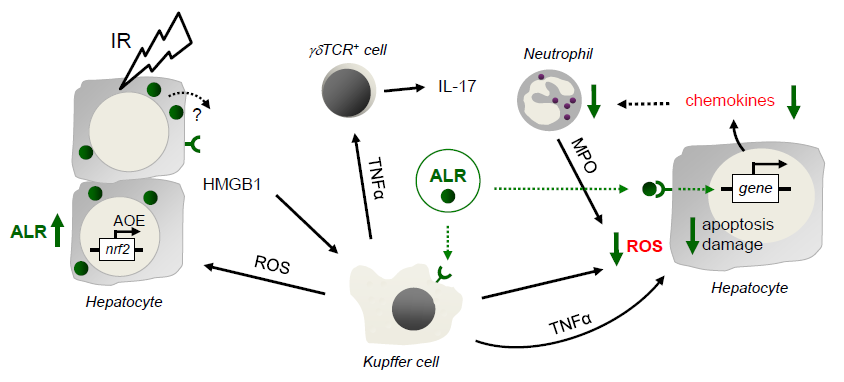
1. Introduction
2. Materials and Methods
2.1. Mouse Liver IRI Model
2.2. Human Liver Biopsies
2.3. Histological Analysis (Hematoxylin-Eosin)
2.4. TUNEL Assay—Analysis of Apoptosis
2.5. Immunohistochemistry
2.6. Isolation of Cells
2.7. In Vitro Culture Conditions
2.8. Flow Cytometry and Cytokine Measurement
2.9. Reactive Oxygen Substances and Oxidative Stress
2.10. Gene Expression (Quantitative Real-Time PCR)
2.11. Western Blot Analysis
2.12. Statistics
3. Results
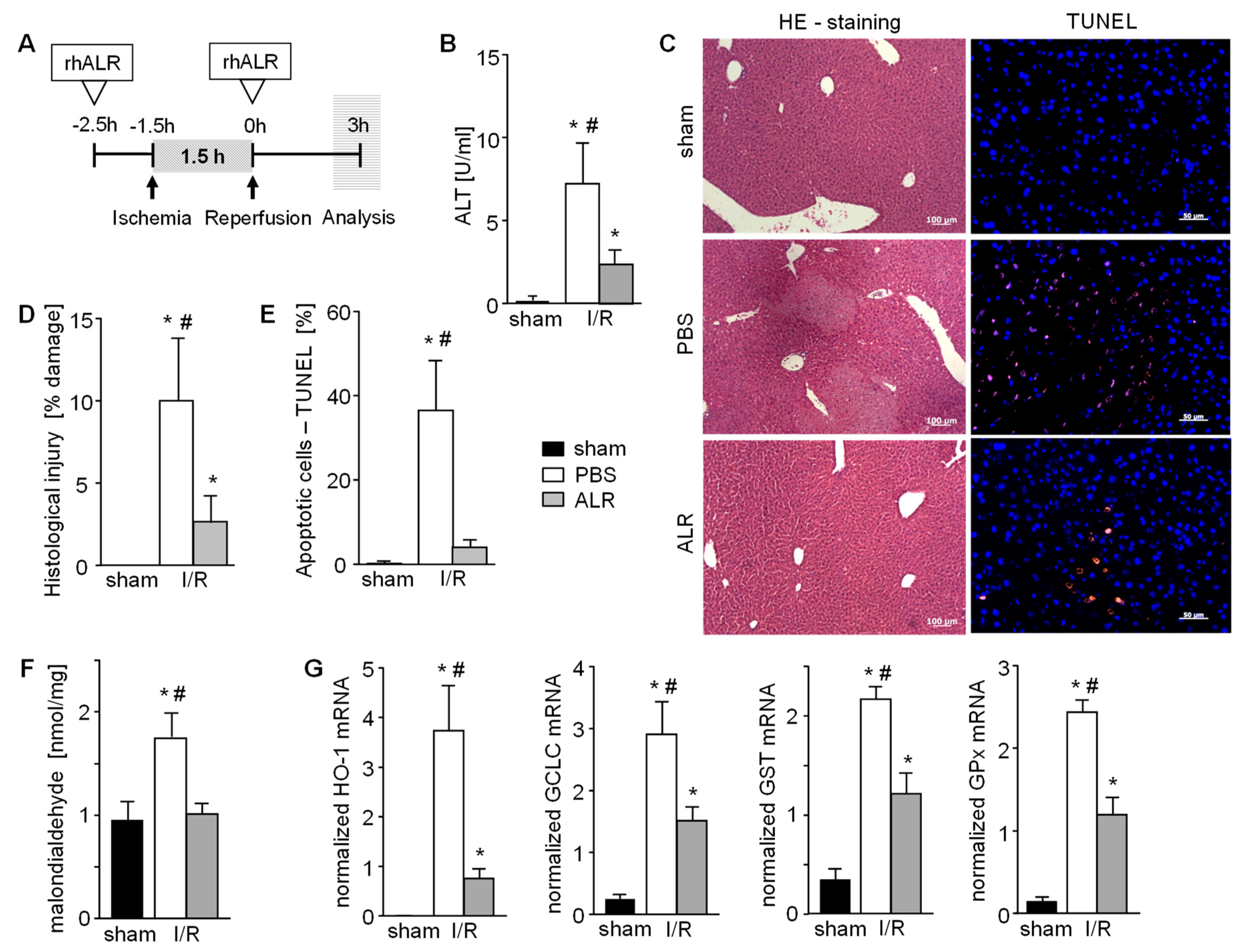
3.1. Treatment with ALR Prevents Hepatic Ischemic Reperfusion Injury While Reducing Tissue Damage and Oxidative Stress
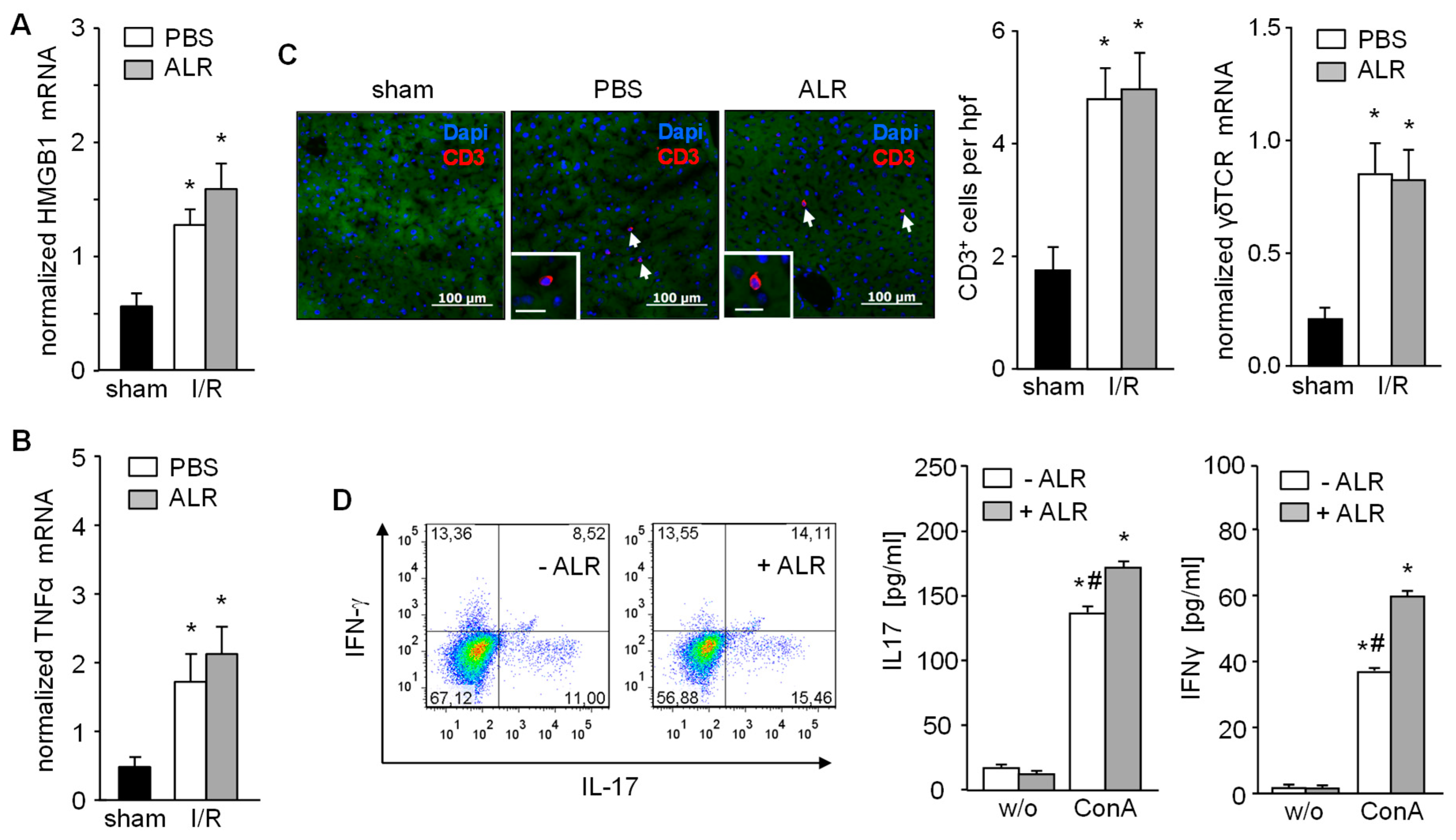
3.2. Recruitment and Activation of γδT Cells (CD3+) Remains Unaffected by ALR Treatment during IR
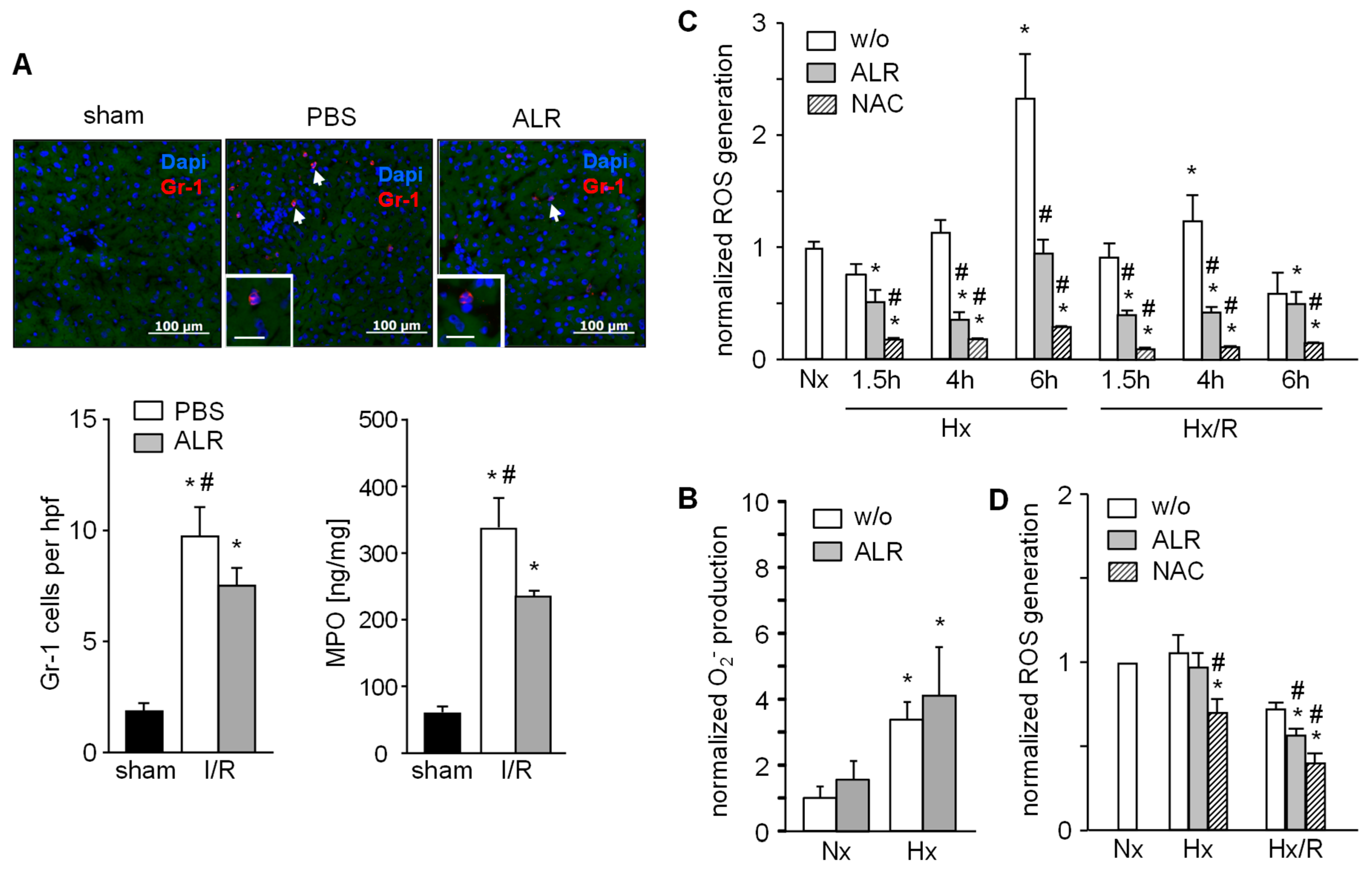
3.3. ALR Attenuates Hepatic Infiltration of Neutrophils (Gr1+ ) and Reduces ROS Generation by Macrophages (RAW 264.7) and Hepatocytes
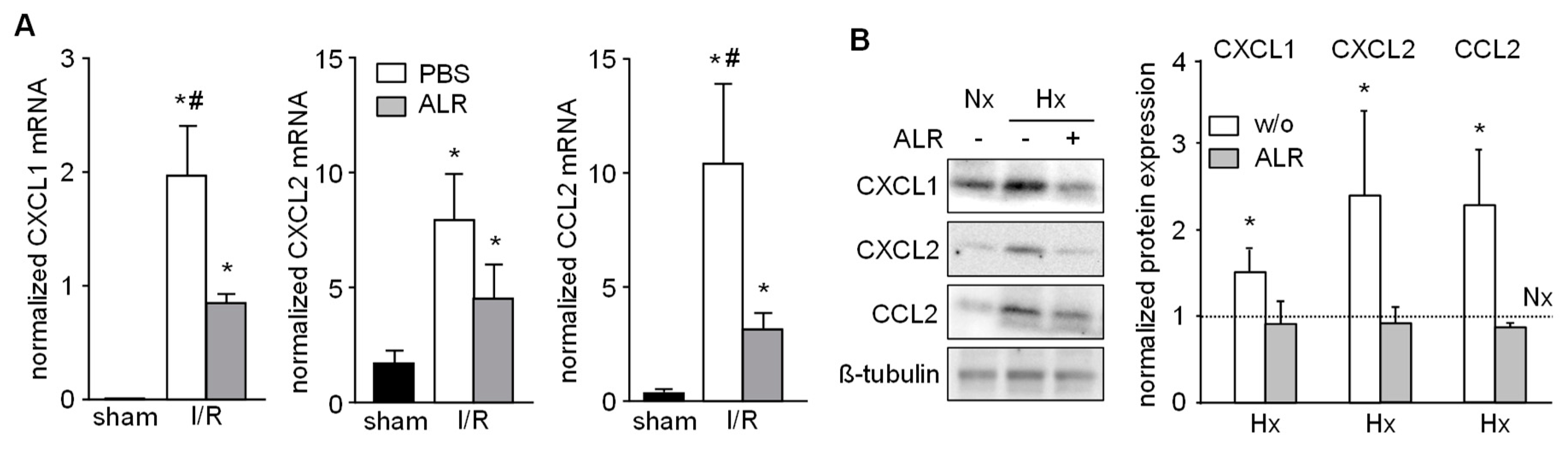
3.4. Expression of Chemo-Attractants in IRI Liver Tissue and Primary Hepatocytes Cells is Mitigated by ALR
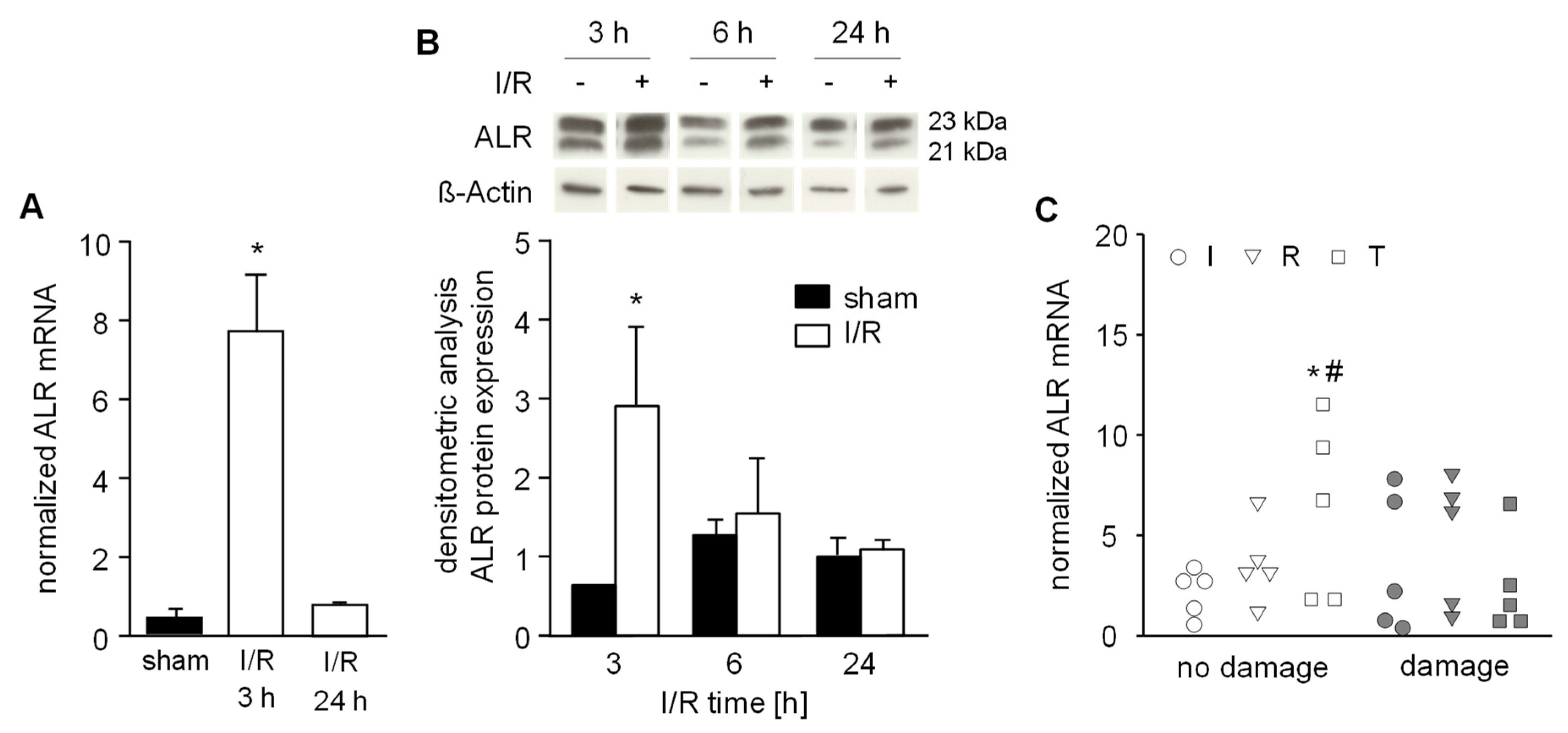
3.5. Expression of ALR is Increased in an IR Mouse Model and in Human Trans-planted Liver Tissue without Injury after Reperfusion
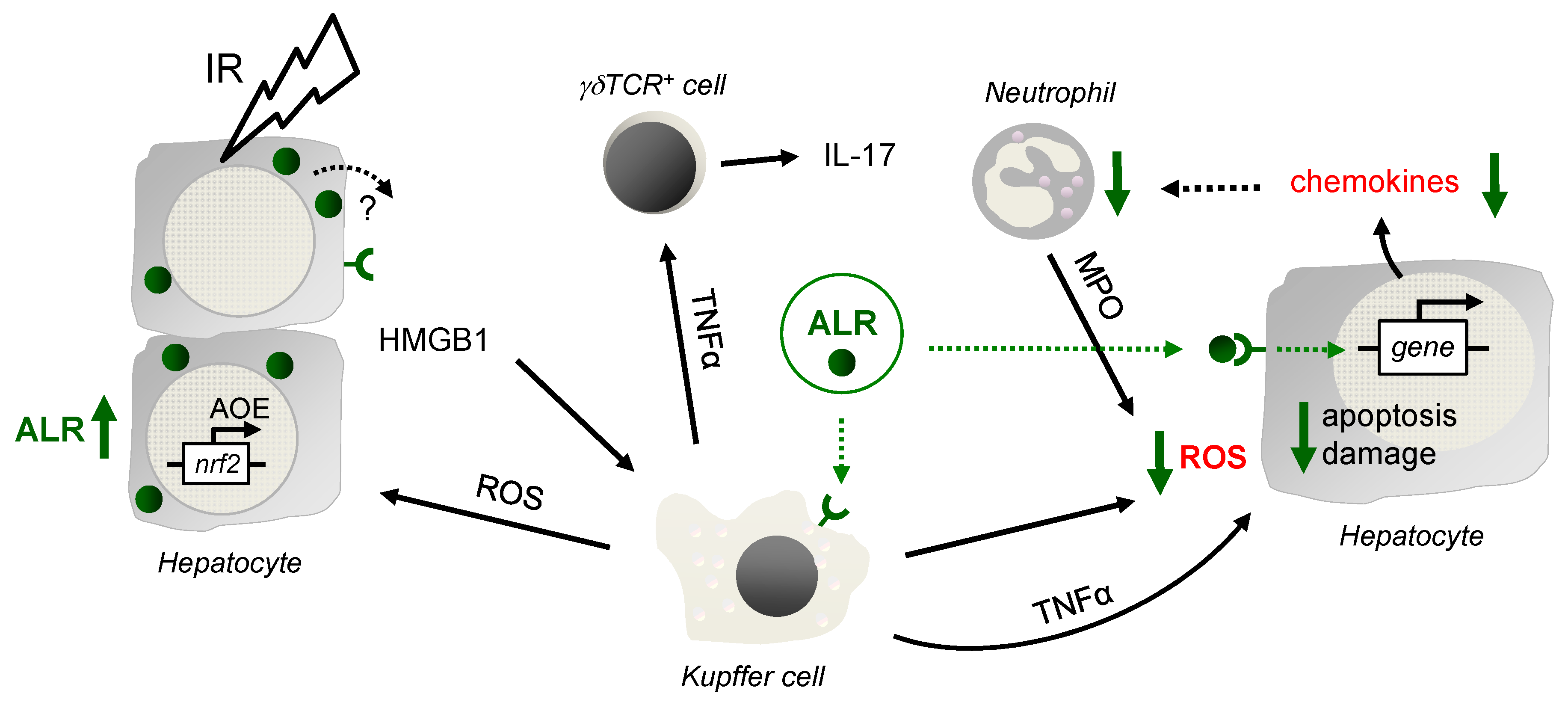
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hubscher, S.G. Transplantation pathology. Semin. Liver Dis. 2009, 29, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Konishi, T.; Lentsch, A.B. Hepatic Ischemia/Reperfusion: Mechanisms of Tissue Injury, Repair, and Regeneration. Gene Expr. 2017, 17, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Woolbright, B.L. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant. Rev. (Orlando) 2012, 26, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. The impact of sterile inflammation in acute liver injury. J. Clin. Transl. Res. 2017, 3, 170–188. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr. Pharmacol. Rep. 2018, 4, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Eggenhofer, E.; Rovira, J.; Sabet-Baktach, M.; Groell, A.; Scherer, M.N.; Dahlke, M.H.; Farkas, S.A.; Loss, M.; Koehl, G.E.; Lang, S.A.; et al. Unconventional RORgammat+ T cells drive hepatic ischemia reperfusion injury. J. Immunol. 2013, 191, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Nakanuma, S.; Ahmed, A.K.; Makino, I.; Hayashi, H.; Oyama, K.; Nakagawara, H.; Tajima, H.; Takamura, H.; Ninomiya, I.; et al. Ischemia reperfusion-facilitated sinusoidal endothelial cell injury in liver transplantation and the resulting impact of extravasated platelet aggregation. Eur. Surg. 2016, 48, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Weiss, T.S. Augmenter of liver regeneration: Essential for growth and beyond. Cytokine Growth Factor Rev. 2019, 45, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.S.; Lupke, M.; Ibrahim, S.; Buechler, C.; Lorenz, J.; Ruemmele, P.; Hofmann, U.; Melter, M.; Dayoub, R. Attenuated lipotoxicity and apoptosis is linked to exogenous and endogenous augmenter of liver regeneration by different pathways. PLoS ONE 2017, 12, e0184282. [Google Scholar] [CrossRef] [PubMed]
- Ilowski, M.; Kleespies, A.; de Toni, E.N.; Donabauer, B.; Jauch, K.W.; Hengstler, J.G.; Thasler, W.E. Augmenter of liver regeneration (ALR) protects human hepatocytes against apoptosis. Biochem. Biophys. Res. Commun. 2011, 404, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Polimeno, L.; Pesetti, B.; Annoscia, E.; Giorgio, F.; Francavilla, R.; Lisowsky, T.; Gentile, A.; Rossi, R.; Bucci, A.; Francavilla, A. Alrp, a survival factor that controls the apoptotic process of regenerating liver after partial hepatectomy in rats. Free Radic. Res. 2011, 45, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Polimeno, L.; Pesetti, B.; De Santis, F.; Resta, L.; Rossi, R.; De Palma, A.; Girardi, B.; Amoruso, A.; Francavilla, A. Decreased expression of the augmenter of liver regeneration results in increased apoptosis and oxidative damage in human-derived glioma cells. Cell Death Dis. 2012, 3, e289. [Google Scholar] [CrossRef] [PubMed]
- Polimeno, L.; Pesetti, B.; Lisowsky, T.; Iannone, F.; Resta, L.; Giorgio, F.; Mallamaci, R.; Buttiglione, M.; Santovito, D.; Vitiello, F.; et al. Protective effect of augmenter of liver regeneration on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic. Res. 2009, 43, 865–875. [Google Scholar] [PubMed]
- Wang, N.; Wang, Z.; Sun, H.; Shi, X.; Zhang, Y.; Liu, Q. Augmenter of liver regeneration improves therapeutic effect of hepatocyte homotransplantation in acute liver failure rats. Int. Immunopharmacol. 2013, 15, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, S.; Long, F.; Li, J.; Gong, J. Augmenter of liver regeneration attenuates acute rejection after rat liver transplantation. Am. J. Surg. 2016, 212, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Li, Y.; Zhang, L.; Xia, N.; Liu, Q.; Sun, H.; Guo, H. Augmenter of liver regeneration attenuates inflammation of renal ischemia/reperfusion injury through the NF-kappa B pathway in rats. Int. Urol. Nephrol. 2015, 47, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Khandoga, A.; Mende, K.; Iskandarov, E.; Rosentreter, D.; Schelcher, C.; Reifart, J.; Jauch, K.W.; Thasler, W.E. Augmenter of liver regeneration attenuates inflammatory response in the postischemic mouse liver in vivo. J. Surg. Res. 2014, 192, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Hines, I.N.; Zibari, G.; Pavlick, K.; Gray, L.; Kitagawa, Y.; Grisham, M.B. Mouse model of liver ischemia and reperfusion injury: Method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic. Biol. Med. 2009, 46, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, R.; Buerger, L.; Ibrahim, S.; Melter, M.; Weiss, T.S. Augmenter of liver regeneration (ALR) exhibits a dual signaling impact on hepatic acute-phase response. Exp. Mol. Pathol. 2017, 102, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Calderone, V.; Cefaro, C.; Ciofi-Baffoni, S.; Gallo, A.; Kallergi, E.; Lionaki, E.; Pozidis, C.; Tokatlidis, K. Molecular recognition and substrate mimicry drive the electron-transfer process between MIA40 and ALR. Proc. Natl. Acad. Sci. USA 2011, 108, 4811–4816. [Google Scholar] [CrossRef] [PubMed]
- Sanders, E.J.; Wride, M.A. Ultrastructural identification of apoptotic nuclei using the TUNEL technique. Histochem. J. 1996, 28, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Dahlke, M.H.; Loi, R.; Warren, A.; Holz, L.; Popp, F.C.; Weiss, D.J.; Piso, P.; Bowen, D.G.; McCaughan, G.W.; Schlitt, H.J.; et al. Immune-mediated hepatitis drives low-level fusion between hepatocytes and adult bone marrow cells. J. Hepatol. 2006, 44, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.S.; Kim, J.W.; Cha, Y.N.; Kim, C. A quantitative nitroblue tetrazolium assay for determining intracellular superoxide anion production in phagocytic cells. J. Immunoass. Immunochem. 2006, 27, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, R.; Vogel, A.; Schuett, J.; Lupke, M.; Spieker, S.M.; Kettern, N.; Hildt, E.; Melter, M.; Weiss, T.S. Nrf2 activates augmenter of liver regeneration (ALR) via antioxidant response element and links oxidative stress to liver regeneration. Mol. Med. 2013, 19, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Petrowsky, H.; Hong, J.C.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, R.H.; Curbishley, S.M.; Weston, C.J.; Adams, D.H.; Afford, S.C. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transpl. 2010, 16, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, A.; Kim, J.S.; Kon, K.; Jaeschke, H.; Ikejima, K.; Watanabe, S.; Lemasters, J.J. Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology 2008, 48, 1644–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, E.; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenes. Tissue Repair 2008, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thasler, W.E.; Dayoub, R.; Muhlbauer, M.; Hellerbrand, C.; Singer, T.; Grabe, A.; Jauch, K.W.; Schlitt, H.J.; Weiss, T.S. Repression of cytochrome P450 activity in human hepatocytes in vitro by a novel hepatotrophic factor, augmenter of liver regeneration. J. Pharmacol. Exp. Ther. 2006, 316, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, H.; Fujii, H.; Ogiku, M.; Hosomura, N.; Amemiya, H.; Tsuchiya, M.; Hara, M. Role of IL-17A in neutrophil recruitment and hepatic injury after warm ischemia-reperfusion mice. J. Immunol. 2011, 187, 4818–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiman, Y.; Friedman, S.L. The role of chemokines in acute liver injury. Front. Physiol. 2012, 3, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colletti, L.M.; Kunkel, S.L.; Walz, A.; Burdick, M.D.; Kunkel, R.G.; Wilke, C.A.; Strieter, R.M. Chemokine expression during hepatic ischemia/reperfusion-induced lung injury in the rat. The role of epithelial neutrophil activating protein. J. Clin. Invest. 1995, 95, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; Prelich, J.; Lagoa, C.; Barclay, D.; Zamora, R.; Murase, N.; Gandhi, C.R. Augmenter of liver regeneration (ALR) is a novel biomarker of hepatocellular stress/inflammation: In vitro, in vivo and in silico studies. Mol. Med. 2013, 18, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, S.; Shin, T.; Huber, N.; Eismann, T.; Galloway, E.; Schuster, R.; Blanchard, J.; Edwards, M.J.; Lentsch, A.B. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 2008, 48, 1213–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Xu, C.; Zhang, F.; Ma, J. Comparative analysis of expression profiles of chemokines, chemokine receptors, and components of signaling pathways mediated by chemokines in eight cell types during rat liver regeneration. Genome 2010, 53, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 173–179. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Marra, F. Chemokines in liver inflammation and fibrosis. Front. Biosci. 2002, 7, 1899–1914. [Google Scholar] [CrossRef]
- Woo, C.W.; Siow, Y.L.; Karmin, O. Homocysteine induces monocyte chemoattractant protein-1 expression in hepatocytes mediated via activator protein-1 activation. J. Biol. Chem. 2008, 283, 1282–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singha, B.; Gatla, H.R.; Vancurova, I. Transcriptional regulation of chemokine expression in ovarian cancer. Biomolecules 2015, 5, 223–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiguchi, N.; Wang, L.; Mukhopadhyay, P.; Park, O.; Jeong, W.I.; Lafdil, F.; Osei-Hyiaman, D.; Moh, A.; Fu, X.Y.; Pacher, P.; et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 2008, 134, 1148–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Li, W.; Jia, X.; An, W. Alleviation of Ischemia-Reperfusion Injury in Liver Steatosis by Augmenter of Liver Regeneration Is Attributed to Antioxidation and Preservation of Mitochondria. Transplantation 2017, 101, 2340–2348. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, T.S.; Lupke, M.; Dayoub, R.; Geissler, E.K.; Schlitt, H.J.; Melter, M.; Eggenhofer, E. Augmenter of Liver Regeneration Reduces Ischemia Reperfusion Injury by Less Chemokine Expression, Gr-1 Infiltration and Oxidative Stress. Cells 2019, 8, 1421. https://doi.org/10.3390/cells8111421
Weiss TS, Lupke M, Dayoub R, Geissler EK, Schlitt HJ, Melter M, Eggenhofer E. Augmenter of Liver Regeneration Reduces Ischemia Reperfusion Injury by Less Chemokine Expression, Gr-1 Infiltration and Oxidative Stress. Cells. 2019; 8(11):1421. https://doi.org/10.3390/cells8111421
Chicago/Turabian StyleWeiss, Thomas S., Madeleine Lupke, Rania Dayoub, Edward K. Geissler, Hans J. Schlitt, Michael Melter, and Elke Eggenhofer. 2019. "Augmenter of Liver Regeneration Reduces Ischemia Reperfusion Injury by Less Chemokine Expression, Gr-1 Infiltration and Oxidative Stress" Cells 8, no. 11: 1421. https://doi.org/10.3390/cells8111421
APA StyleWeiss, T. S., Lupke, M., Dayoub, R., Geissler, E. K., Schlitt, H. J., Melter, M., & Eggenhofer, E. (2019). Augmenter of Liver Regeneration Reduces Ischemia Reperfusion Injury by Less Chemokine Expression, Gr-1 Infiltration and Oxidative Stress. Cells, 8(11), 1421. https://doi.org/10.3390/cells8111421