Myocardial Adaptation in Pseudohypoxia: Signaling and Regulation of mPTP via Mitochondrial Connexin 43 and Cardiolipin

{kind=link}
Abstract
:1. Introduction
2. Cardioprotection and Mitochondrial Energetics
3. Cardiac Mitochondrial Energetics in Partial Oxygen Deprivation
4. Metabolic Preconditioning
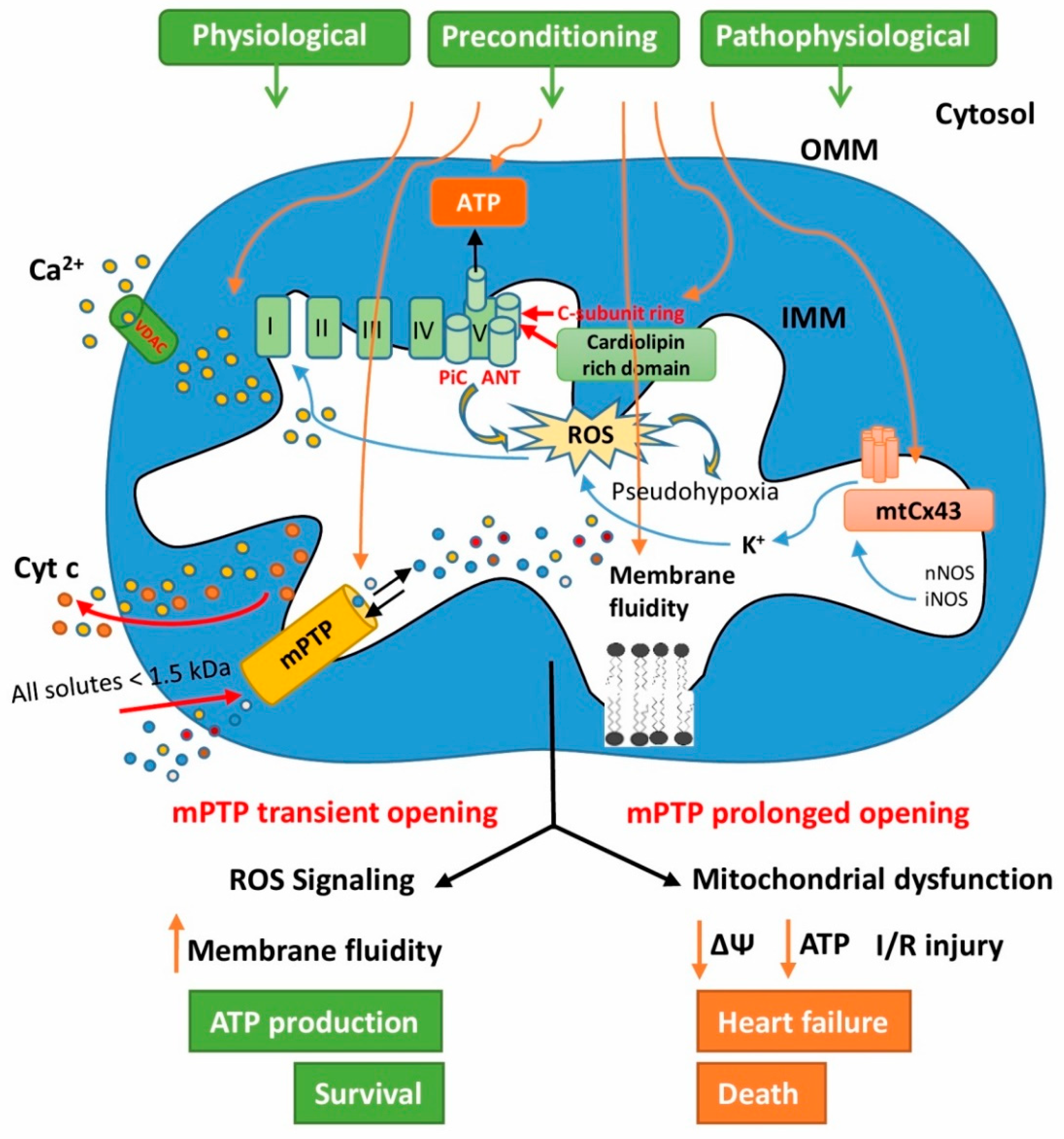
5. Involvement of Mitochondrial Connexin 43 in Cardioprotection
6. The Role of Cardiolipin in Heart Mitochondrial Signaling
7. The Role of Mitochondrial Permeability Transition Pores in Signaling Processes of Cardioprotection
8. Unregulated Mitochondrial Permeability Transition Pore Opening
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Karch, J.; Molkentin, J.D. Regulated necrotic cell death. Circ. Res. 2015, 116, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases—From pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Meerson, F.Z.; Malyshev, I.Y.; Zamotrinsky, A.V. Adaptive protection of the heart and stabilization of myocardial structures. Basic Res. Cardiol. 1991, 86, 87–98. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Ferree, A.; Shirihai, O. Mitochondrial dynamics: The intersection of form and function. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2012; Volume 748, pp. 13–40. [Google Scholar]
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An energetic view of stress: Focus on mitochondria. Front. Neuroendocrinol. 2018, 49, 72–85. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Schulz, R.; Baxter, G.F. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol. Rev. 2007, 59, 418–458. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Perrelli, M.-G. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186. [Google Scholar] [CrossRef]
- Giaccia, A.J. The biology of hypoxia: The role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004, 18, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: Molecular and pharmacological aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 2009, 45, 643–650. [Google Scholar] [CrossRef]
- Assaly, R.; de Tassigny, A.D.A.; Paradis, S.; Jacquin, S.; Berdeaux, A.; Morin, D. Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia–reoxygenation in adult cardiomyocytes. Eur. J. Pharmacol. 2012, 675, 6–14. [Google Scholar] [CrossRef]
- Gadicherla, A.K.; Wang, N.; Bulic, M.; Agullo-Pascual, E.; Lissoni, A.; De Smet, M.; Delmar, M.; Bultynck, G.; Krysko, D.V.; Camara, A.; et al. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 2017, 112, 27. [Google Scholar] [CrossRef]
- Ardehali, A.; Ports, T.A. Myocardial Oxygen Supply and Demand* *Cardiovascular Research Institute, and Division of Cardiology, Department of Medicine, University of California, San Francisco. Chest 1990, 98, 699–705. [Google Scholar] [CrossRef]
- Manneschi, L.; Federico, A. Polarographic analyses of subsarcolemmal and intermyofibrillar mitochondria from rat skeletal and cardiac muscle. J. Neurol. Sci. 1995, 128, 151–156. [Google Scholar] [CrossRef]
- Kueh, H.Y.; Niethammer, P.; Mitchison, T.J. Maintenance of mitochondrial oxygen homeostasis by cosubstrate compensation. Biophys. J. 2013, 104, 1338–1348. [Google Scholar] [CrossRef]
- Riva, A.; Tandler, B.; Loffredo, F.; Vazquez, E.; Hoppel, C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am. J. Physiol. Circ. Physiol. 2005, 289, H868–H872. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Barth, E. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Ferrari, R.; Cargnoni, A.; Ceconi, C. Anti-ischaemic effect of ivabradine. Pharmacol. Res. 2006, 53, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 48–61. [Google Scholar] [CrossRef]
- Saks, V.; Favier, R.; Guzun, R.; Schlattner, U.; Wallimann, T. Molecular system bioenergetics: Regulation of substrate supply in response to heart energy demands. J. Physiol. 2006, 577, 769–777. [Google Scholar] [CrossRef]
- Kojić, Z.; Kojić, Z.; Kojić, Z.; Šćepanović, L.; Šćepanović, L.; Šćepanović, L.; Popović, N.; Popović, N.; Popović, N. Myocardial oxygen consumption regulation in isolated mouse heart: Assessment by intracoronary administration of exogenous nitric oxide. Acta Physiol. Hung. 2006, 93, 263–270. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1360–1372. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef]
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet contributions to myocardial ischemia/reperfusion injury. Front. Immunol. 2019, 10, 1260. [Google Scholar] [CrossRef]
- Cerychova, R.; Pavlinkova, G. HIF-1, metabolism, and diabetes in the embryonic and adult heart. Front. Endocrinol. 2018, 9, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyengaard, J.R.; Ido, Y.; Kilo, C.; Williamson, J.R. Interactions between hyperglycemia and hypoxia: Implications for diabetic retinopathy. Diabetes 2004, 53, 2931–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegelhöffer, A.; Waczulíková, I.; Ferko, M.; Kincelová, D.; Ziegelhöffer, B.; Ravingerová, T.; Cagalinec, M.; Schönburg, M.; Ziegelhoeffer, T.; Šikurová, L.; et al. Calcium signaling-mediated endogenous protection of cell energetics in the acutely diabetic myocardiumThis article is one of a selection of papers published in a special issue on Advances in Cardiovascular Research. Can. J. Physiol. Pharmacol. 2009, 87, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forini, F.; Nicolini, G.; Iervasi, G. Mitochondria as key targets of cardioprotection in cardiac ischemic disease: Role of thyroid hormone triiodothyronine. Int. J. Mol. Sci. 2015, 16, 6312–6336. [Google Scholar] [CrossRef] [Green Version]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and Ischemia/Reperfusion Injury. Ann. N. Y. Acad. Sci. 2005, 1047, 248–258. [Google Scholar] [CrossRef]
- Ravingerová, T.; Pancza, D.; Ziegelhoffer, A.; Styk, J. Preconditioning modulates susceptibility to ischemia-induced arrhythmias in the rat heart: The role of α-adrenergic stimulation and K(ATP) channels. Physiol. Res. 2002, 51, 101–119. [Google Scholar]
- Waldow, T.; Alexiou, K.; Witt, W.; Albrecht, S.; Wagner, F.; Knaut, M.; Matschke, K. Protection against acute porcine lung ischemia/reperfusion injury by systemic preconditioning via hind limb ischemia. Transpl. Int. 2005, 18, 198–205. [Google Scholar] [CrossRef]
- Chen, Q.; Camara, A.K.S.; Stowe, D.F.; Hoppel, C.L.; Lesnefsky, E.J. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Physiol. Physiol. 2007, 292, C137–C147. [Google Scholar] [CrossRef] [Green Version]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Kharbanda, R.K.; Peters, M.; Walton, B.; Kattenhorn, M.; Mullen, M.; Klein, N.; Vallance, P.; Deanfield, J.; MacAllister, R. Ischemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia-reperfusion in humans In Vivo. Circulation 2001, 103, 1624–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravingerova, T.; Farkasova, V.; Griecsova, L.; Carnicka, S.; Murarikova, M.; Barlaka, E.; Kolar, F.; Bartekova, M.; Lonek, L.; Slezak, J.; et al. Remote preconditioning as a novel “ conditioning” approach to repair the broken heart: Potential mechanisms and clinical applications. Physiol. Res. 2016, 65, S55–S64. [Google Scholar] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Remote ischaemic preconditioning: Underlying mechanisms and clinical application. Cardiovasc. Res. 2008, 79, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D.Y. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef] [PubMed]
- Consolini, A.E.; Ragone, M.I.; Bonazzola, P.; Colareda, G.A. Mitochondrial bioenergetics during ischemia and reperfusion. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2017; Volume 982, pp. 141–167. [Google Scholar]
- Liu, Y.; Silverstein, F.S.; Skoff, R.; Barks, J.D.E. Hypoxic-ischemic oligodendroglial injury in neonatal rat brain. Pediatr. Res. 2002, 51, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Navarro, A.; Boveris, A. The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Physiol. 2007, 292, C670–C686. [Google Scholar] [CrossRef]
- Peterson, L.R.; McKenzie, C.R.; Schaffer, J.E. Diabetic cardiovascular disease: Getting to the heart of the matter. J. Cardiovasc. Transl. Res. 2012, 5, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Holness, M.J. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E855–E862. [Google Scholar] [CrossRef] [Green Version]
- Heather, L.C.; Clarke, K. Metabolism, hypoxia and the diabetic heart. J. Mol. Cell. Cardiol. 2011, 50, 598–605. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1333–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Huang, H.; McElfresh, T.A.; Prosdocimo, D.A.; Stanley, W.C. Impact of anaerobic glycolysis and oxidative substrate selection on contractile function and mechanical efficiency during moderate severity ischemia. Am. J. Physiol. Circ. Physiol. 2008, 295, H939–H945. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, D.L.; Ravussin, E. The role of mitochondria in health and disease. Curr. Opin. Pharmacol. 2009, 9, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, N.H. Pyruvate dehydrogenase kinases: Therapeutic targets for diabetes and cancers. Diabetes Metab. J. 2015, 39, 188. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.; Lindsay, J.G. Molecular architecture of the pyruvate dehydrogenase complex: Bridging the gap. Biochem. Soc. Trans. 2006, 34, 815–818. [Google Scholar] [CrossRef] [Green Version]
- Jeoung, N.H.; Harris, C.R.; Harris, R.A. Regulation of pyruvate metabolism in metabolic-related diseases. Rev. Endocr. Metab. Disord. 2014, 15, 99–110. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Dodd, M.S.; da Sousa Fialho, M.L.; Montes Aparicio, C.N.; Kerr, M.; Timm, K.N.; Griffin, J.L.; Luiken, J.J.F.P.; Glatz, J.F.C.; Tyler, D.J.; Heather, L.C. Fatty acids prevent hypoxia-inducible Factor-1α signaling through decreased succinate in diabetes. JACC Basic Transl. Sci. 2018, 3, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Wittig, R.; Coy, J.F. The role of glucose metabolism and glucose-associated signalling in cancer. Perspect. Medicin. Chem. 2007, 1. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Gunaydin, B.; Çakici, I.; Soncul, H.; Kalaycioglu, S.; Çevik, C.; Sancak, B.; Kanzik, I.; Karadenizli, Y. Does remote organ ischemia trigger cardiac preconditioning during coronary artery surgery? Pharmacol. Res. 2000, 41, 493–496. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Daşu, A.; Toma-Daşu, I.; Karlsson, M. Theoretical simulation of tumour oxygenation and results from acute and chronic hypoxia. Phys. Med. Biol. 2003, 48, 2829–2842. [Google Scholar] [PubMed] [Green Version]
- Yan, L.-J.; Wu, J.; Jin, Z.; Zheng, H. Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes Metab. Syndr. Obes. Targets Ther. 2016, 9, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, J.R.; Chang, K.; Frangos, M.; Hasan, K.S.; Ido, Y.; Kawamura, T.; Nyengaard, J.R.; van Den Enden, M.; Kilo, C.; Tilton, R.G. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993, 42, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsang, T.M.; Teahan, O.; et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 2008, 283, 22700–22708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.J.; Harris, R.A.; Wu, P.; Pehleman, T.L.; Heigenhauser, G.J.F.; Spriet, L.L. Human skeletal muscle PDH kinase activity and isoform expression during a 3-day high-fat/low-carbohydrate diet. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1151–E1158. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeon, J.-H.; Min, B.-K.; Ha, C.-M.; Thoudam, T.; Park, B.-Y.; Lee, I.-K. Role of the pyruvate dehydrogenase complex in metabolic remodeling: Differential pyruvate dehydrogenase complex functions in metabolism. Diabetes Metab. J. 2018, 42, 270. [Google Scholar] [CrossRef]
- Lee, I.-K. The role of pyruvate dehydrogenase kinase in diabetes and obesity. Diabetes Metab. J. 2014, 38, 181. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Liu, Q.; Leng, J.; Zheng, Y.; Li, J. The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases. Life Sci. 2015, 121, 97–103. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, S.-G.; Belghiti, J. Liver transplantation for HCC: Its role. J. Hepatobiliary Pancreat. Sci. 2010, 17, 443–448. [Google Scholar] [CrossRef]
- Ham, P.B.; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, C.; de Souza Junior, A.L.; Carbonaro, M.; Bolsoni-Lopes, A.; Figueroa, D.; de Souza, L.E.; Silva, K.A.S.; Consolim-Colombo, F.; Curi, R.; Irigoyen, M.C. Glucose and fatty acid metabolism in infarcted heart from streptozotocin-induced diabetic rats after 2 weeks of tissue remodeling. Cardiovasc. Diabetol. 2015, 14, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Takashi, E.; Kudo, M.; Ishiwata, T.; Naito, Z. Contradictory effects of short-and long-term hyperglycemias on ischemic injury of myocardium via intracellular signaling pathway. Exp. Mol. Pathol. 2004, 76, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ferko, M.; Habodászová, D.; Waczulíková, I.; Mujkošová, J.; Kucharská, J.; Šikurová, L.; Ziegelhoffer, B.; Styk, J.; Ziegelhffer, A. Endogenous protective mechanisms in remodeling of rat heart mitochondrial membranes in the acute phase of streptozotocin-induced diabetes. Physiol. Res. 2008, 57, S67–S73. [Google Scholar] [PubMed]
- Zhu, X.-H.; Yuan, H.-J.; Wu, Y.-N.; Kang, Y.; Jiao, J.-J.; Gao, W.-Z.; Liu, Y.-X.; Lou, J.-S.; Xia, Z. Non-invasive limb ischemic pre-conditioning reduces oxidative stress and attenuates myocardium ischemia-reperfusion injury in diabetic rats. Free Radic. Res. 2011, 45, 201–210. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.F. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, T.; Kasuga, M.; Akanuma, Y.; Ezaki, O.; Takaku, F. Decreased autophosphorylation of the insulin receptor-kinase in streptozotocin-diabetic rats. J. Biol. Chem. 1984, 259, 14208–14216. [Google Scholar]
- Ziegelhöffer, A.; Ravingerová, T.; Styk, J.; Tribulová, N.; Volkovová, K.; Šeboková, J.; Breier, A. Diabetic cardiomyopathy in rats: Biochemical mechanisms of increased tolerance to calcium overload. Diabetes Res. Clin. Pract. 1996, 31, S93–S103. [Google Scholar] [CrossRef]
- Oliveira, P.J.; Seiça, R.; Coxito, P.M.; Rolo, A.P.; Palmeira, C.M.; Santos, M.S.; Moreno, A.J.M. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003, 554, 511–514. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, B.; Figueroa, D.M.T.; Fang, J.; Rosa, K.T.; Llesuy, S.; De Angelis, K.; Irigoyen, M.C. Short-term diabetes attenuates left ventricular dysfunction and mortality rates after myocardial infarction in rodents. Clinics 2011, 66, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Malfitano, C.; Barboza, C.A.; Mostarda, C.; da Palma, R.K.; dos Santos, C.P.; Rodrigues, B.; Freitas, S.C.F.; Belló-Klein, A.; Llesuy, S.; Irigoyen, M.-C.; et al. Diabetic hyperglycemia attenuates sympathetic dysfunction and oxidative stress after myocardial infarction in rats. Cardiovasc. Diabetol. 2014, 13, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, M.; Neely, J.R. Hearts from diabetic rats are more resistant to in vitro ischemia: Possible role of altered Ca2+ metabolism. Circ. Res. 1988, 62, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kancirová, I.; Jašová, M.; Muráriková, M.; Sumbalová, Z.; Uličná, O.; Ravingerová, T.; Waczulíková, I.; Ziegelhöffer, A.; Ferko, M. Cardioprotection induced by remote ischemic preconditioning preserves the mitochondrial respiratory function in acute diabetic myocardium. Physiol. Res. 2016, 65, S611–S619. [Google Scholar] [PubMed]
- Ferko, M.; Gvozdjaková, A.; Kucharská, J.; Mujkošová, J.; Waczulíková, I.; Styk, J.; Ravingerová, T.; Ziegelhöffer-Mihalovičová, B.; Ziegelhöffer, A. Functional remodeling of heart mitochondria in acute diabetes: Interrelationships between damage endogenous protection and adaptation. Gen. Physiol. Biophys. 2006, 25, 397–413. [Google Scholar]
- Mapanga, R.F.; Rajamani, U.; Dlamini, N.; Zungu-Edmondson, M.; Kelly-Laubscher, R.; Shafiullah, M.; Wahab, A.; Hasan, M.Y.; Fahim, M.A.; Rondeau, P.; et al. Oleanolic acid: A novel cardioprotective agent that blunts hyperglycemia-induced contractile dysfunction. PLoS ONE 2012, 7, e47322. [Google Scholar] [CrossRef] [Green Version]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Tribulova, N.; Szeiffova Bacova, B.; Egan Benova, T.; Knezl, V.; Barancik, M.; Slezak, J. Omega-3 index and anti-arrhythmic potential of omega-3 PUFAs. Nutrients 2017, 9, 1191. [Google Scholar] [CrossRef] [Green Version]
- Katengua-Thamahane, E.; Szeiffova Bacova, B.; Bernatova, I.; Sykora, M.; Knezl, V.; Van Rooyen, J.; Tribulova, N. Effects of red palm oil on myocardial antioxidant enzymes, nitric oxide synthase and heart function in spontaneously hypertensive rats. Int. J. Mol. Sci. 2017, 18, 2476. [Google Scholar] [CrossRef] [Green Version]
- Sykora, M.; Szeiffova Bacova, B.; Egan Benova, T.; Barancik, M.; Zurmanova, J.; Rauchova, H.; Weismann, P.; Pavelka, S.; Kurahara, L.H.; Slezak, J.; et al. Cardiac Cx43 and ECM Responses to Altered Thyroid Status Are Blunted in Spontaneously Hypertensive versus Normotensive Rats. Int. J. Mol. Sci. 2019, 20, 3758. [Google Scholar] [CrossRef] [Green Version]
- Prado, N.J.; Egan Beňová, T.; Diez, E.R.; Knezl, V.; Lipták, B.; Ponce Zumino, A.Z.; Llamedo-Soria, M.; Szeiffová Bačová, B.; Miatello, R.M.; Tribulová, N. Melatonin receptor activation protects against low potassium-induced ventricular fibrillation by preserving action potentials and connexin-43 topology in isolated rat hearts. J. Pineal Res. 2019, 67. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, R.D.; Wang, H.-Z.; Beblo, D.A.; Chilton, M.G.; Harris, A.L.; Beyer, E.C.; Brink, P.R. Selectivity of connexin-specific gap junctions does not correlate with channel conductance. Circ. Res. 1995, 77, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, D.A. Connexins, connexons, and intercellular communication. Annu. Rev. Biochem. 1996, 65, 475–502. [Google Scholar] [CrossRef] [PubMed]
- Severs, N. Gap junction alterations in human cardiac disease. Cardiovasc. Res. 2004, 62, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Sáez, J.C.; Retamal, M.A.; Basilio, D.; Bukauskas, F.F.; Bennett, M.V.L. Connexin-based gap junction hemichannels: Gating mechanisms. Biochim. Biophys. Acta Biomembr. 2005, 1711, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Dodoni, G.; Rodriguezsinovas, A.; Cabestrero, A.; Ruizmeana, M.; Gres, P.; Konietzka, I.; Lopeziglesias, C.; Garciadorado, D.; Dilisa, F. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Cabestrero, A.; Boengler, K.; Heusch, G.; Garcia-Dorado, D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2007, 77, 325–333. [Google Scholar] [CrossRef]
- Boengler, K.; Stahlhofen, S.; Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef]
- Rodríguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; García-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta Biomembr. 2018, 1860, 174–181. [Google Scholar] [CrossRef]
- Boengler, K.; Schulz, R. Connexin 43 and mitochondria in cardiovascular health and disease. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2017; Volume 982, pp. 227–246. [Google Scholar]
- Boengler, K.; Ruiz-Meana, M.; Gent, S.; Ungefug, E.; Soetkamp, D.; Miro-Casas, E.; Cabestrero, A.; Fernandez-Sanz, C.; Semenzato, M.; Di Lisa, F.; et al. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J. Cell. Mol. Med. 2012, 16, 1649–1655. [Google Scholar] [CrossRef]
- Denuc, A.; Núñez, E.; Calvo, E.; Loureiro, M.; Miro-Casas, E.; Guarás, A.; Vázquez, J.; Garcia-Dorado, D. New protein-protein interactions of mitochondrial connexin 43 in mouse heart. J. Cell. Mol. Med. 2016, 20, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Si, R.; Scott, B.T.; Makino, A. Mitochondrial connexin40 regulates mitochondrial calcium uptake in coronary endothelial cells. Am. J. Physiol. Physiol. 2017, 312, C398–C406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, R.; Görge, P.M.; Görbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, F.R.; Luo, Y.; Li, X.; Boengler, K.; Buechert, A.; García-Dorado, D.; Di Lisa, F.; Schulz, R.; Heusch, G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ. Res. 2005, 97, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Korge, P.; Honda, H.M.; Weiss, J.N. Protection of cardiac mitochondria by diazoxide and protein kinase C: Implications for ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2002, 99, 3312–3317. [Google Scholar] [CrossRef] [Green Version]
- Kirca, M.; Kleinbongard, P.; Soetkamp, D.; Heger, J.; Csonka, C.; Ferdinandy, P.; Schulz, R. Interaction between Connexin 43 and nitric oxide synthase in mice heart mitochondria. J. Cell. Mol. Med. 2015, 19, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Soetkamp, D.; Nguyen, T.T.; Menazza, S.; Hirschhäuser, C.; Hendgen-Cotta, U.B.; Rassaf, T.; Schlüter, K.D.; Boengler, K.; Murphy, E.; Schulz, R. S-nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res. Cardiol. 2014, 109, 433. [Google Scholar] [CrossRef] [Green Version]
- Katoh, H.; Nishigaki, N.; Hayashi, H. Diazoxide opens the mitochondrial permeability transition pore and alters Ca2+ transients in rat ventricular myocytes. Circulation 2002, 105, 2666–2671. [Google Scholar] [CrossRef] [Green Version]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.S.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miró, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90–dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the heart of mitochondrial metabolism. Cell. Mol. Life Sci. 2008, 65, 2493–2506. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Unsay, J.D.; Cosentino, K.; Subburaj, Y.; García-Sáez, A.J. Cardiolipin effects on membrane structure and dynamics. Langmuir 2013, 29, 15878–15887. [Google Scholar] [CrossRef]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef]
- Ferko, M.; Farkasova, V.; Jasova, M.; Kancirova, I.; Ravingerova, T.; Adameova, A.D.; Andelova, N.; Waczulikova, I. Hypercholesterolemia antagonized heart adaptation and functional remodeling of the mitochondria observed in acute diabetes mellitus subjected to ischemia/reperfusion injury. J. Physiol. Pharmacol. 2018, 69, 685–697. [Google Scholar]
- Palovicova, V.; Bardelcikova, A.; Obernauerova, M. Absence of anionic phospholipids in Kluyveromyces lactis cells is fatal without F1-catalysed ATP hydrolysis. Can. J. Microbiol. 2012, 58, 694–702. [Google Scholar] [CrossRef]
- Jones, R.M.; Bagchi, M.; Das, D.K. Preconditioning of heart by repeated stunning: Adaptive modification of myocardial lipid membrane. Basic Res. Cardiol. 1992, 87, 527–535. [Google Scholar] [CrossRef]
- Muráriková, M.; Ferko, M.; Waczulíková, I.; Jašová, M.; Kancirová, I.; Murínová, J.; Ravingerová, T. Changes in mitochondrial properties may contribute to enhanced resistance to ischemia–reperfusion injury in the diabetic rat heart. Can. J. Physiol. Pharmacol. 2017, 95, 969–976. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bakovic, M. Formation and regulation of mitochondrial membranes. Int. J. Cell Biol. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Slabe, T.J.; Stoll, M.S.K.; Minkler, P.E.; Hoppel, C.L. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am. J. Physiol. Hear Circ. Physiol. 2001, 280, H2770–H2778. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in mitochondrial complex i activity in ischemic/reperfused rat heart. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.G.; Cheng, P.; Angel, A.; Hatch, G.M. Thyroxine stimulates phosphatidylglycerolphosphate synthase activity in rat heart mitochondria. Biochim. Biophys. Acta Lipids Lipid Metab. 1995, 1256, 241–244. [Google Scholar] [CrossRef]
- Hostetler, K.Y. Effect of thyroxine on the activity of mitochondrial cardiolipin synthase in rat liver. Biochim. Biophys. Acta Lipids Lipid Metab. 1991, 1086, 139–140. [Google Scholar] [CrossRef]
- He, Q.; Han, X. Cardiolipin remodeling in diabetic heart. Chem. Phys. Lipids 2014, 179, 75–81. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial metabolism in aging heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1544–1554. [Google Scholar] [CrossRef]
- Yin, H.; Zhu, M. Free radical oxidation of cardiolipin: Chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radic. Res. 2012, 46, 959–974. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Mikhaeil, J.S.; Leveille, C.F.; Saint, C.; LeBlanc, P.J. Cardiolipin content, linoleic acid composition, and tafazzin expression in response to skeletal muscle overload and unload stimuli. Sci. Rep. 2017, 7, 2060. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J. Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, V.A.; Greenberg, M.L. Cardiolipin-mediated cellular signaling. Adv. Exp. Med. Biol. 2013, 991, 195–213. [Google Scholar] [PubMed]
- Sparagna, G.C.; Lesnefsky, E.J. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 2009, 53, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together. J. Biol. Chem. 2002, 277, 43553–43556. [Google Scholar] [CrossRef] [Green Version]
- Gomez, B.; Robinson, N.C. Phospholipase digestion of bound cardiolipin reversibly inactivates bovine cytochrome bc 1 †. Biochemistry 1999, 38, 9031–9038. [Google Scholar] [CrossRef]
- Petrosillo, G.; Ruggiero, F.M.; Di Venosa, N.; Paradies, G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: Role of reactive oxygen species and cardiolipin. FASEB J. 2003, 7, 714–716. [Google Scholar] [CrossRef]
- Sedlák, E.; Robinson, N.C. Phospholipase A 2 digestion of cardiolipin bound to bovine cytochrome c oxidase alters both activity and quaternary structure †. Biochemistry 1999, 38, 14966–14972. [Google Scholar] [CrossRef]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [Green Version]
- Pestana, C.R.; Silva, C.H.T.P.; Pardo-Andreu, G.L.; Rodrigues, F.P.; Santos, A.C.; Uyemura, S.A.; Curti, C. Ca2+ binding to c-state of adenine nucleotide translocase (ANT)-surrounding cardiolipins enhances (ANT)-Cys56 relative mobility: A computational-based mitochondrial permeability transition study. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 176–182. [Google Scholar] [CrossRef]
- Ostrander, D.B.; Sparagna, G.C.; Amoscato, A.A.; McMillin, J.B.; Dowhan, W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J. Biol. Chem. 2001, 276, 38061–38067. [Google Scholar]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.; Zhivotovsky, B.; Orrenius, S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007, 14, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Smith Eble, K.; Coleman, W.B.; Hantgan, R.R.; Cunningham, C.C. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31 P nuclear magnetic resonance spectroscopy. J. Biol. Chem. 1990, 265, 19434–19440. [Google Scholar]
- Duncan, A.L.; Robinson, A.J.; Walker, J.E. Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl. Acad. Sci. USA 2016, 113, 8687–8692. [Google Scholar] [CrossRef] [Green Version]
- Biasutto, L.; Azzolini, M.; Szabò, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2515–2530. [Google Scholar] [CrossRef]
- Carraro, M.; Checchetto, V.; Szabó, I.; Bernardi, P. F-ATP synthase and the permeability transition pore: Fewer doubts, more certainties. FEBS Lett. 2019, 593, 1542–1553. [Google Scholar] [CrossRef] [Green Version]
- Jašová, M.; Kancirová, I.; Waczulíková, I.; Ferko, M. Mitochondria as a target of cardioprotection in models of preconditioning. J. Bioenerg. Biomembr. 2017, 49, 357–368. [Google Scholar] [CrossRef]
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharmacol. Exp. Ther. 2009, 330, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. The mitochondrial megachannel is the permeability transition pore. J. Bioenerg. Biomembr. 1992, 24, 111–117. [Google Scholar] [CrossRef]
- Javadov, S.; Karmazyn, M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell. Physiol. Biochem. 2007, 20, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nazari, A.; Sadr, S.S.; Faghihi, M.; Azizi, Y.; Hosseini, M.-J.; Mobarra, N.; Tavakoli, A.; Imani, A. Vasopressin attenuates ischemia–reperfusion injury via reduction of oxidative stress and inhibition of mitochondrial permeability transition pore opening in rat hearts. Eur. J. Pharmacol. 2015, 760, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Argaud, L. Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc. Res. 2004, 61, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ziegelhöffer-Mihalovičová, B.; Waczulíková, I.; Šikurová, L.; Styk, J.; Čársky, J.; Ziegelhöffer, A. Remodelling of the sarcolemma in diabetic rat hearts: The role of membrane fluidity. Mol. Cell. Biochem. 2003, 249, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Naderi, R.; Imani, A.; Faghihi, M.; Moghimian, M. Phenylephrine induces early and late cardioprotection through mitochondrial permeability transition pore in the isolated rat heart. J. Surg. Res. 2010, 164, e37–e42. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic functions of cyclophilin d and the mitochondrial permeability transition pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Mnatsakanyan, N.; Beutner, G.; Porter, G.A.; Alavian, K.N.; Jonas, E.A. Physiological roles of the mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Quintanilla, R.A. Development or disease: Duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Parodi-Rullán, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia–reperfusion: Whether cyclophilin D is a viable target for cardioprotection? Cell. Mol. Life Sci. 2017, 74, 2795–2813. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Ghosh, P.; Wan, R.; Ouyang, X.; Cheng, H.; Mattson, M.P.; Cheng, A. Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1−42 on neural progenitor cell proliferation. Neurobiol. Aging 2014, 35, 975–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ. Res. 2016, 118, 834–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegelhoffer-Mihalovicova, B.; Okruhlicová, L.; Tribulová̈, N.; Ravingerová, T.; Volkovová, K.; Šeboková, J.; Ziegelhöffer, A. Mitochondrial contact sites detected by creatine phosphokinase activity in the hearts of normal and diabetic rats: Is mitochondrial contact sites formation a calcium-dependent process? Gen. Physiol. Biophys. 1997, 16, 329–338. [Google Scholar]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. The mitochondrial permeability transition pore and cancer: Molecular mechanisms involved in cell death. Front. Oncol. 2014, 4, 302. [Google Scholar] [CrossRef] [Green Version]
- Heather, L.C.; Cole, M.A.; Tan, J.-J.; Ambrose, L.J.A.; Pope, S.; Abd-Jamil, A.H.; Carter, E.E.; Dodd, M.S.; Yeoh, K.K.; Schofield, C.J.; et al. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res. Cardiol. 2012, 107, 268. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Why F-ATP Synthase remains a strong candidate as the mitochondrial permeability transition pore. Front. Physiol. 2018, 9, 1543. [Google Scholar] [CrossRef] [Green Version]
- Karch, J.; Molkentin, J.D. Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10396–10397. [Google Scholar] [CrossRef] [Green Version]
- Beutner, G.; Alanzalon, R.E.; Porter, G.A. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci. Rep. 2017, 7, 14488. [Google Scholar] [CrossRef] [Green Version]
- Claypool, S.M. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 12816–12821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [Green Version]
- Šileikytė, J.; Forte, M. The mitochondrial permeability transition in mitochondrial disorders. Oxidative Med. Cell. Longev. 2019, 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Perrelli, M.-G.; Pagliaro, P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: Therapeutic implications. Antioxid. Redox Signal. 2013, 18, 556–599. [Google Scholar] [CrossRef] [PubMed]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D promotes brain mitochondrial F1FO ATP synthase dysfunction in aging mice. J. Alzheimer Dis. 2016, 55, 1351–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griecsová, L.; Farkašová, V.; Gáblovskỳ, I.; Khandelwal, V.K.M.; Bernátová, I.; Tatarková, Z.; Kaplan, P.; Ravingerová, T. Effect of maturation on the resistance of rat hearts against ischemia. Study of potential molecular mechanisms. Physiol. Res. 2015, 64, S685–S696. [Google Scholar]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Protection by cyclosporin a of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef]
- Abeti, R.; Abramov, A.Y. Mitochondrial Ca2+ in neurodegenerative disorders. Pharmacol. Res. 2015, 99, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Walters, A.M.; Porter, G.A.; Brookes, P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 2012, 111, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, G.; Duchen, M.R. Investigating the mitochondrial permeability transition pore in disease phenotypes and drug screening. Curr. Protoc. Pharmacol. 2019, 85, e59. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferko, M.; Andelová, N.; Szeiffová Bačová, B.; Jašová, M. Myocardial Adaptation in Pseudohypoxia: Signaling and Regulation of mPTP via Mitochondrial Connexin 43 and Cardiolipin. Cells 2019, 8, 1449. https://doi.org/10.3390/cells8111449
Ferko M, Andelová N, Szeiffová Bačová B, Jašová M. Myocardial Adaptation in Pseudohypoxia: Signaling and Regulation of mPTP via Mitochondrial Connexin 43 and Cardiolipin. Cells. 2019; 8(11):1449. https://doi.org/10.3390/cells8111449
Chicago/Turabian StyleFerko, Miroslav, Natália Andelová, Barbara Szeiffová Bačová, and Magdaléna Jašová. 2019. "Myocardial Adaptation in Pseudohypoxia: Signaling and Regulation of mPTP via Mitochondrial Connexin 43 and Cardiolipin" Cells 8, no. 11: 1449. https://doi.org/10.3390/cells8111449
APA StyleFerko, M., Andelová, N., Szeiffová Bačová, B., & Jašová, M. (2019). Myocardial Adaptation in Pseudohypoxia: Signaling and Regulation of mPTP via Mitochondrial Connexin 43 and Cardiolipin. Cells, 8(11), 1449. https://doi.org/10.3390/cells8111449