The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System


{kind=link}
Abstract
:1. Neutrophils in the Central Nervous System (CNS)
2. Neutrophil Extracellular Traps (NETs) in Physiology and Pathology
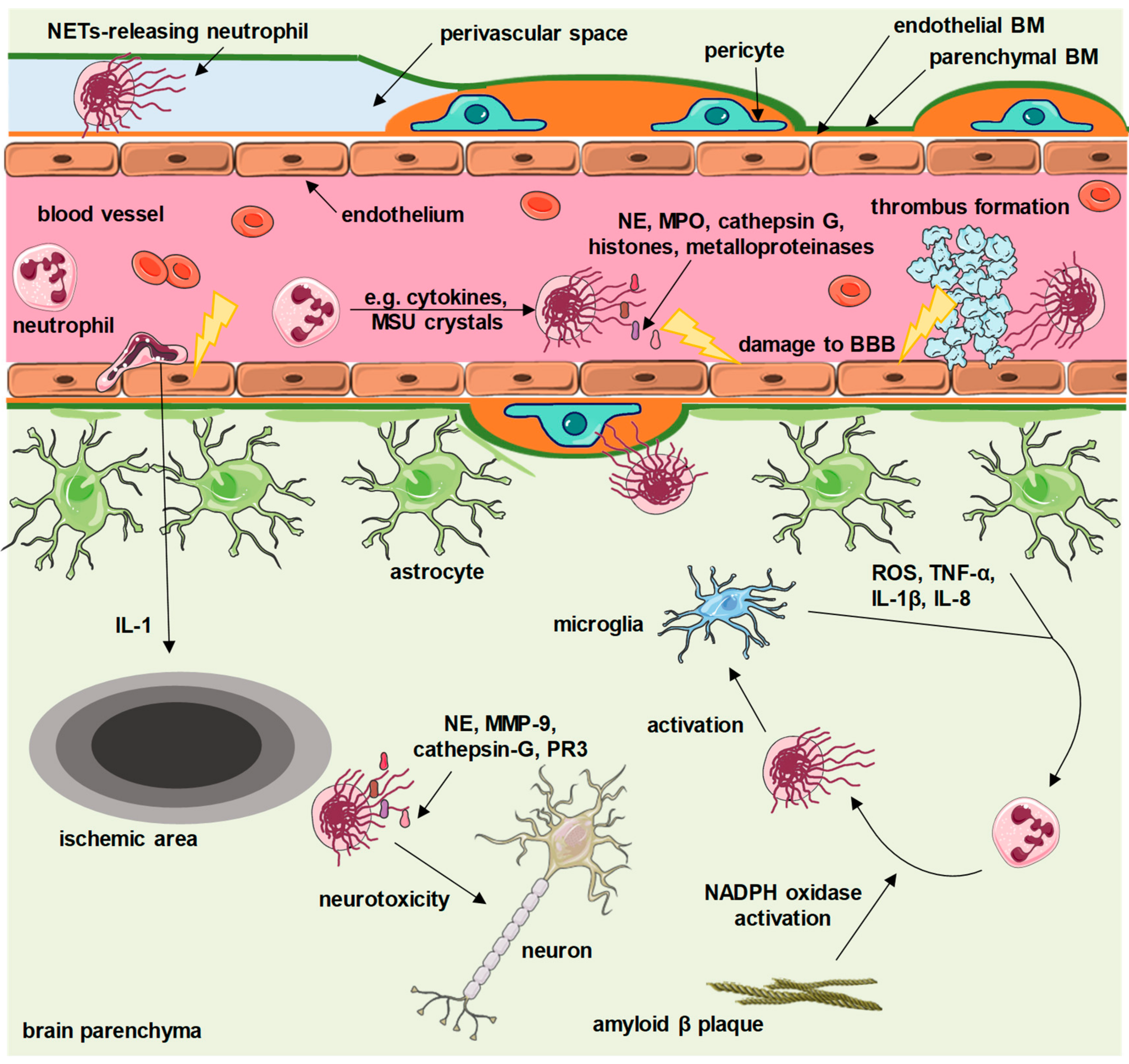
3. NETs in Ischemic Stroke
4. Neurodegeneration
5. Autoimmune Diseases
6. CNS Infections
7. Peripheral Diseases with Infiltration of Central Nervous System by Neutrophils
8. May NETs Play a Role in the Development of Brain Tumors?
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perry, V.H.; Anthony, D.C.; Bolton, S.J.; Brown, H.C. The blood-brain barrier and the inflammatory response. Mol. Med. Today 1997, 3, 335–341. [Google Scholar] [CrossRef]
- Gemechu, M.J.; Bentivoglio, M. T Cell recruitment in the brain during normal aging. Front. Cell Neurosci. 2012, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Onodera, H.; Shiga, Y.; Nakamura, M.; Ninomiya, M.; Kihara, T.; Kogure, K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat: Effects of neutrophil depletion. Stroke 1994, 25, 1469–1475. [Google Scholar] [CrossRef]
- Strecker, K.J.; Schmidt, A.; Schabitz, W.R.; Minnerup, J. Neutrophil granulocytes in cerebral ischemia—Evolution from killers to key players. Neurochem. Int. 2017, 107, 117–126. [Google Scholar] [CrossRef]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET: Current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef]
- Manda, A.; Pruchniak, M.P.; Arazna, M.; Demkow, U. Neutrophil extracellular traps in physiology and pathology. Cent. Eur. J. Immunol. 2014, 39, 116–121. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Menegazzi, R.; Decleva, E.; Dri, P. Killing by neutrophil extracellular traps: Fact or folklore? Blood 2012, 119, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic cancer-induced neutrophil extracellular traps: A potential contributor to cancer-associated thrombosis. Int. J. Mol. Sci. 2017, 18, 487. [Google Scholar] [CrossRef] [PubMed]
- Bryk, A.H.; Prior, S.M.; Plens, K.; Konieczynska, M.; Hohendorff, J.; Malecki, M.T.; Butenas, S.; Undas, A. Predictors of neutrophil extracellular traps markers in type 2 diabetes mellitus: Associations with a prothrombotic state and hypofibrinolysis. Cardiovasc. Diabetol. 2019, 18, 49. [Google Scholar] [CrossRef] [PubMed]
- Skrzeczynska-Moncznik, J.; Wlodarczyk, A.; Zabieglo, K.; Kapinska-Mrowiecka, M.; Marewicz, E.; Dubin, A.; Potempa, J.; Cichy, J. Secretory leukocyte proteinase inhibitor-competent DNA deposits are potent stimulators of plasmacytoid dendritic cells: Implication for psoriasis. J. Immunol. 2012, 189, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm. 2017, 14, 29. [Google Scholar] [CrossRef]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef]
- Zhang, R.L.; Chopp, M.; Chen, H.; Garcia, J.H. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2H) middle cerebral artery occlusion in the rat. J. Neurol. Sci. 1994, 125, 3–10. [Google Scholar] [CrossRef]
- Garcia, J.H.; Liu, K.F.; Yoshida, Y.; Lian, J.; Chen, S.; del Zoppo, G.J. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am. J. Pathol. 1994, 144, 188–199. [Google Scholar]
- Chu, H.X.; Kim, H.A.; Lee, S.; Moore, J.P.; Chan, C.T.; Vinh, A.; Gelderblom, M.; Arumugam, T.V.; Broughton, B.R.; Drummond, G.R.; et al. Immune cell infiltration in malignant middle cerebral artery infarction: Comparison with transient cerebral ischemia. J. Cereb. Blood. Flow. Metab. 2014, 34, 450–459. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef] [PubMed]
- Perez-de-Puig, I.; Miro-Mur, F.; Ferrer-Ferrer, M.; Gelpi, E.; Pedragosa, J.; Justicia, C.; Urra, X.; Chamorro, A.; Planas, A.M. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015, 129, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Humphreys, N.; Lane, T.E.; Grencis, R.; Rothwell, N. Chronic systemic infection exacerbates ischemic brain damage via a CCL5 (regulated on activation, normal T-cell expressed and secreted)-mediated proinflammatory response in mice. J. Neurosci. 2010, 30, 10086–10095. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.; van Eijk, M.; Haagsman, H.P.; Hartshorn, K.L. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol. 2016, 11, 441–453. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Laridan, E.; Denorme, F.; Desender, L.; Francois, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; de Meyer, S.F. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef]
- Ruhnau, J.; Schulze, K.; Gaida, B.; Langner, S.; Kessler, C.; Broker, B.; Dressel, A.; Vogelgesang, A. Stroke alters respiratory burst in neutrophils and monocytes. Stroke 2014, 45, 794–800. [Google Scholar] [CrossRef]
- Tillack, K.; Breiden, P.; Martin, R.; Sospedra, M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 2012, 188, 3150–3159. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.J.; Iqbal, M.; Loh, T.; Trusko, S.P.; Scott, R.; Siman, R. Cathepsin G: Localization in human cerebral cortex and generation of amyloidogenic fragments from the beta-amyloid precursor protein. Neuroscience 1994, 60, 607–619. [Google Scholar] [CrossRef]
- Czirr, E.; Wyss-Coray, T. The immunology of neurodegeneration. J. Clin. Invest. 2012, 122, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat. Med. 2006, 12, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Szekely, C.A.; Zandi, P.P. Non-Steroidal anti-inflammatory drugs and Alzheimer’s disease: The epidemiological evidence. CNS Neurol. Disord. Drug Targets 2010, 9, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Pietronigro, E.C.; della Bianca, V.; Zenaro, E.; Constantin, G. NETosis in Alzheimer’s Disease. Front. Immunol. 2017, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimers Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef]
- Rodrigues, S.F.; Granger, D.N. Blood cells and endothelial barrier function. Tissue Barriers 2015, 3, e978720. [Google Scholar] [CrossRef] [Green Version]
- Suttorp, N.; Nolte, A.; Wilke, A.; Drenckhahn, D. Human neutrophil elastase increases permeability of cultured pulmonary endothelial cell monolayers. Int. J. Microcirc. Clin. Exp. 1993, 13, 187–203. [Google Scholar]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; el Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Duchen, M.R. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos. Trans. R Soc. Lond. B Biol. Sci. 2005, 360, 2309–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allam, R.; Kumar, S.V.; Darisipudi, M.N.; Anders, H.J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 2014, 92, 465–472. [Google Scholar] [CrossRef]
- Peppin, G.J.; Weiss, S.J. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc. Natl. Acad. Sci. USA 1986, 83, 4322–4326. [Google Scholar] [CrossRef] [Green Version]
- Gilthorpe, J.D.; Oozeer, F.; Nash, J.; Calvo, M.; Bennett, D.L.; Lumsden, A.; Pini, A. Extracellular histone H1 is neurotoxic and drives a pro-inflammatory response in microglia. F1000Research 2013, 2, 148. [Google Scholar] [CrossRef]
- Yoshii, F.; Shinohara, Y. Autoimmune neurological diseases. J. Jpn. Med. Assoc. 2004, 47, 425–430. [Google Scholar]
- Tillack, K.; Naegele, M.; Haueis, C.; Schippling, S.; Wandinger, K.P.; Martin, R.; Sospedra, M. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J. Neuroimmunol. 2013, 261, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Croxford, A.L.; Kurschus, F.C.; Waisman, A. Mouse models for multiple sclerosis: Historical facts and future implications. Biochim. Biophys. Acta 2011, 1812, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Afraei, S.; Sedaghat, R.; Zavareh, F.T.; Aghazadeh, Z.; Ekhtiari, P.; Azizi, G.; Mirshafiey, A. Therapeutic effects of pegylated-interferon-alpha2a in a mouse model of multiple sclerosis. Cent. Eur. J. Immunol. 2018, 43, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Hambro, C.A.; Johnston, A.; Stuart, P.E.; Tsoi, L.C.; Nair, R.P.; Elder, J.T. Neutrophil extracellular traps induce human Th17 cells: Effect of psoriasis-associated TRAF3IP2 genotype. J. Invest. Derm. 2019, 139, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation: Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, M.; Dzopalic, T.; Zivanovic, S.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I.; Vojinovic, S.; Marjanovic, G.; Savic, V.; Colic, M. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand. J. Immunol. 2014, 79, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2016, 136, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. Neuroreport 2018, 29, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-Related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Woodberry, T.; Bouffler, S.E.; Wilson, A.S.; Buckland, R.L.; Brustle, A. The emerging role of neutrophil granulocytes in multiple sclerosis. J. Clin. Med. 2018, 7, 511. [Google Scholar] [CrossRef] [Green Version]
- MS Research Australia. NETS: A New Target for Treating MS. Available online: https://msra.org.au/project/neutrophil-extracellular-traps-new-target-treating-ms/ (accessed on 10 October 2019).
- Tay, S.H.; Mak, A. Anti-NR2A/B antibodies and other major molecular mechanisms in the pathogenesis of cognitive dysfunction in systemic lupus erythematosus. Int. J. Mol. Sci. 2015, 16, 10281–10300. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Kawasaki, T.; Shigematsu, K.; Kawamura, K.; Oka, N. Neutrophil extracellular traps in neuropathy with anti-neutrophil cytoplasmic autoantibody-associated microscopic polyangiitis. Clin. Rheumatol. 2017, 36, 913–917. [Google Scholar] [CrossRef]
- Conly, J.M.; Ronald, A.R. Cerebrospinal fluid as a diagnostic body fluid. Am. J. Med. 1983, 75, 102–108. [Google Scholar] [CrossRef]
- Hutchings, M.; Weller, R.O. Anatomical relationships of the pia mater to cerebral blood vessels in man. J. Neurosurg. 1986, 65, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Malipiero, U.; Koedel, U.; Pfister, H.W.; Leveen, P.; Burki, K.; Reith, W.; Fontana, A. TGFbeta receptor II gene deletion in leucocytes prevents cerebral vasculitis in bacterial meningitis. Brain 2006, 129, 2404–2415. [Google Scholar] [CrossRef] [PubMed]
- De Buhr, N.; Reuner, F.; Neumann, A.; Stump-Guthier, C.; Tenenbaum, T.; Schroten, H.; Ishikawa, H.; Muller, K.; Beineke, A.; Hennig-Pauka, I.; et al. Neutrophil extracellular trap formation in the Streptococcus suis-infected cerebrospinal fluid compartment. Cell Microbiol. 2017, 19. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, T.; Fisher, J.; Bakochi, A.; Neumann, A.; Cardoso, J.F.P.; Karlsson, C.A.Q.; Pavan, C.; Lundgaard, I.; Nilson, B.; Reinstrup, P.; et al. Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nat. Commun. 2019, 10, 1667. [Google Scholar] [CrossRef] [PubMed]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.J.; Ayvazian, J.H.; Tillett, W.S. Crystalline pancreatic desoxyribonuclease as an adjunct to the treatment of pneumococcal meningitis. N. Engl. J. Med. 1959, 260, 893–900. [Google Scholar] [CrossRef]
- Jhelum, H.; Sori, H.; Sehgal, D. A novel extracellular vesicle-associated endodeoxyribonuclease helps Streptococcus pneumoniae evade neutrophil extracellular traps and is required for full virulence. Sci. Rep. 2018, 8, 7985. [Google Scholar] [CrossRef]
- Storisteanu, D.M.; Pocock, J.M.; Cowburn, A.S.; Juss, J.K.; Nadesalingam, A.; Nizet, V.; Chilvers, E.R. Evasion of neutrophil extracellular traps by respiratory pathogens. Am. J. Respir. Cell. Mol. Biol. 2017, 56, 423–431. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Berends, E.T.M.; Chan, R.; Schwab, E.; Roy, S.; Sen, C.K.; Torres, V.J.; Wozniak, D.J. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc. Natl. Acad. Sci. USA 2018, 115, 7416–7421. [Google Scholar] [CrossRef] [Green Version]
- Alhamdi, Y.; Toh, C.H. Recent advances in pathophysiology of disseminated intravascular coagulation: The role of circulating histones and neutrophil extracellular traps. F1000Research 2017, 6, 2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.W.; Li, S.; Dai, S.S. Neutrophils in traumatic brain injury (TBI): Friend or foe? J. Neuroinflamm. 2018, 15, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Buhr, N.; Neumann, A.; Jerjomiceva, N.; von Kockritz-Blickwede, M.; Baums, C.G. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology 2014, 160, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Buhr, N.; Stehr, M.; Neumann, A.; Naim, H.Y.; Valentin-Weigand, P.; von Kockritz-Blickwede, M.; Baums, C.G. Identification of a novel DNase of Streptococcus suis (EndAsuis) important for neutrophil extracellular trap degradation during exponential growth. Microbiology 2015, 161, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Boeltz, S.; Munoz, L.E.; Fuchs, T.A.; Herrmann, M. Neutrophil extracellular traps open the Pandora’s box in severe malaria. Front. Immunol. 2017, 8, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorn, C.; Janko, C.; Latzko, M.; Chaurio, R.; Schett, G.; Herrmann, M. Monosodium urate crystals induce extracellular DNA traps in neutrophils, eosinophils, and basophils but not in mononuclear cells. Front. Immunol. 2012, 3, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorn, C.; Janko, C.; Krenn, V.; Zhao, Y.; Munoz, L.E.; Schett, G.; Herrmann, M. Bonding the foe—NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front. Immunol. 2012, 3, 376. [Google Scholar] [CrossRef] [Green Version]
- Baker, V.S.; Imade, G.E.; Molta, N.B.; Tawde, P.; Pam, S.D.; Obadofin, M.O.; Sagay, S.A.; Egah, D.Z.; Iya, D.; Afolabi, B.B.; et al. Cytokine-Associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar. J. 2008, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R. Cerebral malaria: Mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr. Res. 2010, 68, 267–274. [Google Scholar] [CrossRef]
- He, H.; Geng, T.; Chen, P.; Wang, M.; Hu, J.; Kang, L.; Song, W.; Tang, H. NK cells promote neutrophil recruitment in the brain during sepsis-induced neuroinflammation. Sci. Rep. 2016, 6, 27711. [Google Scholar] [CrossRef]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Mello, C.; Riazi, K.; Le, T.; Stevens, K.M.; Wang, A.; McKay, D.M.; Pittman, Q.J.; Swain, M.G. P-selectin-mediated monocyte-cerebral endothelium adhesive interactions link peripheral organ inflammation to sickness behaviors. J. Neurosci. 2013, 33, 14878–14888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Olson, B.; Michaelis, K.A.; Torres, E.R.S.; Patel, E.M.; Jeng, S.; McWeeney, S.; et al. A distinct neutrophil population invades the central nervous system during pancreatic cancer. bioRxiv 2019, 659060. [Google Scholar] [CrossRef]
- Miller, A.H.; Ancoli-Israel, S.; Bower, J.E.; Capuron, L.; Irwin, M.R. Neuroendocrine-immune mechanisms of behavioral comorbidities in patients with cancer. J. Clin. Oncol. 2008, 26, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Meyers, C.A. Neurocognitive dysfunction in cancer patients. Oncology 2000, 14, 75–79. [Google Scholar]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer. Cell. 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Schulz, M.; Salamero-Boix, A.; Niesel, K.; Alekseeva, T.; Sevenich, L. Microenvironmental regulation of tumor progression and therapeutic response in brain metastasis. Front. Immunol. 2019, 10, 1713. [Google Scholar] [CrossRef]
- Uehara, T.; Baba, I.; Nomura, Y. Induction of cytokine-induced neutrophil chemoattractant in response to various stresses in rat C6 glioma cells. Brain Res. 1998, 790, 284–292. [Google Scholar] [CrossRef]
- Fossati, G.; Ricevuti, G.; Edwards, S.W.; Walker, C.; Dalton, A.; Rossi, M.L. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999, 98, 349–354. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.H.; Wang, Q.H.; Elakkad, A.; et al. Glioblastoma-Infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, E.F.; Herzig, A.; Kruger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; Bernuth, H.V.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Lang, S.; Brandau, S. Modulation of neutrophil granulocytes in the tumor microenvironment: Mechanisms and consequences for tumor progression. Semin. Cancer Biol. 2013, 23, 141–148. [Google Scholar] [CrossRef]
- Shamamian, P.; Schwartz, J.D.; Pocock, B.J.Z.; Monea, S.; Whiting, D.; Marcus, S.G.; Mignatti, P. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: A role for inflammatory cells in tumor invasion and angiogenesis. J. Cell Physiol. 2001, 189, 197–206. [Google Scholar] [CrossRef]
- Wu, C.F.; Andzinski, L.; Kasnitz, N.; Kroger, A.; Klawonn, F.; Lienenklaus, S.; Weiss, S.; Jablonska, J. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int. J. Cancer 2015, 137, 837–847. [Google Scholar] [CrossRef]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Ferri, L.E. Neutrophils promote liver metastasis via mac-1-mediated interactions with circulating tumor cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bambury, R.M.; Teo, M.Y.; Power, D.G.; Yusuf, A.; Murray, S.; Battley, J.E.; Drake, C.; O’Dea, P.; Bermingham, N.; Keohane, C.; et al. The association of pre-treatment neutrophil to lymphocyte ratio with overall survival in patients with glioblastoma multiforme. J. Neurooncol. 2013, 114, 149–154. [Google Scholar] [CrossRef] [PubMed]
- McNamara, G.M.; Templeton, A.J.; Maganti, M.; Walter, T.; Horgan, A.M.; McKeever, L.; Min, T.; Amir, E.; Knox, J.J. Neutrophil/lymphocyte ratio as a prognostic factor in biliary tract cancer. Eur. J. Cancer. 2014, 50, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, K.; Nakasu, Y.; Kurakane, T.; Hayashi, N.; Harada, H.; Nozaki, K. Elevated preoperative neutrophil-to-lymphocyte ratio as a predictor of worse survival after resection in patients with brain metastasis. J. Neurosurg. 2017, 127, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manda-Handzlik, A.; Demkow, U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells 2019, 8, 1477. https://doi.org/10.3390/cells8121477
Manda-Handzlik A, Demkow U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells. 2019; 8(12):1477. https://doi.org/10.3390/cells8121477
Chicago/Turabian StyleManda-Handzlik, Aneta, and Urszula Demkow. 2019. "The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System" Cells 8, no. 12: 1477. https://doi.org/10.3390/cells8121477
APA StyleManda-Handzlik, A., & Demkow, U. (2019). The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells, 8(12), 1477. https://doi.org/10.3390/cells8121477