Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma


Abstract
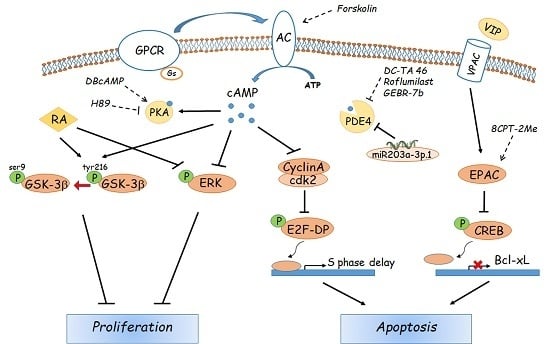
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Physiologic Effectors of cAMP in Hepatocytes
3. Pharmacological Modulators of the cAMP Pathway
4. Role of cAMP Signalling in Non-Hepatic Tumours
5. Role of Cyclic AMP in Hepatocellular Carcinoma
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Couri, T.; Pillai, A. Goals and targets for personalized therapy for HCC. Hepatol. Int. 2019, 13, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Y.; Ye, S.L.; Liu, Y.K.; Qin, L.X.; Sun, H.C.; Ye, Q.H.; Wang, L.; Zhou, J.; Qiu, S.J.; Li, Y.; et al. A decade’s studies on metastasis of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2004, 130, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Blume, H.E. Molecular therapy and prevention of hepatocellular carcinoma. Hepatobiliary Pancreat. Dis. Int. 2003, 2, 11–22. [Google Scholar]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, S2–S6. [Google Scholar] [CrossRef]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Huang, X.Y.; Huang, Z.L.; Zhang, P.B.; Huang, X.Y.; Huang, J.; Wang, H.C.; Xu, B.; Zhou, J.; Tang, Z.Y. CircRNA-100338 Is Associated With mTOR Signaling Pathway and Poor Prognosis in Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 392. [Google Scholar] [CrossRef]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib-chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256–265. [Google Scholar] [CrossRef]
- Riggle, K.M.; Turnham, R.; Scott, J.D.; Yeung, R.S.; Riehl, K.J. Fibrolamellar Hepatocellular Carcinoma: Mechanistic Distinction From Adult Hepatocellular Carcinoma. Pediatr. Blood Cancer 2016, 63, 1163–1167. [Google Scholar] [CrossRef]
- Turnham, R.E.; Smith, F.D.; Kenerson, H.L.; Omar, M.H.; Golkowski, M.; Garcia, I.; Bauer, R.; Lau, H.T.; Sullivan, K.M.; Langeberg, L.K.; et al. An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. eLife 2019, 8, e44187. [Google Scholar] [CrossRef] [PubMed]
- Savai, R.; Pullamsetti, S.S.; Banat, G.A.; Weissmann, N.; Ghofrani, H.A.; Grimminger, F.; Schermuly, R.T. Targeting cancer with phosphodiesterase inhibitors. Expert. Opin. Investig. Drugs 2010, 19, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Massimi, M.; Cardarelli, S.; Galli, F.; Giardi, M.F.; Ragusa, F.; Panera, N.; Cinque, B.; Cifone, M.G.; Biagioni, S.; Giorgi, M. Increase of intracellular cyclic AMP by PDE4 inhibitors affects HepG2 cell cycle progression and survival. J. Cell. Biochem. 2017, 118, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Gong, J.; Jin, Y.; Zhou, Y.; Tong, R.; Wei, X.; Bai, L.; Shi, J. Inhibitors of phosphodiesterase as cancer therapeutics. Eur. J. Med. Chem. 2018, 150, 742–756. [Google Scholar] [CrossRef]
- Ragusa, F.; Panera, N.; Cardarelli, S.; Caioni, G.; Alisi, A.; Giorgi, M.; Massimi, M. Type 4 phosphodiesterases: A possible role in hepatocyte transformation. Eur. J. Histochem. 2018, 62, 7. [Google Scholar]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Zippin, J.H.; Chen, Y.; Straub, S.G.; Hess, K.C.; Diaz, A.; Lee, D.; Tso, P.; Holz, G.G.; Sharp, G.W.; Levin, L.R.; et al. CO2/HCO3(-)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J. Biol. Chem. 2013, 288, 33283–33291. [Google Scholar] [CrossRef]
- Reggi, E.; Diviani, D. The role of A-kinase anchoring proteins in cancer development. Cell Signal. 2017, 40, 143–155. [Google Scholar] [CrossRef]
- Pilkis, S.J.; Claus, T.H.; el-Maghrabi, M.R. The role of cyclic AMP in rapid and long-term regulation of gluconeogenesis and glycolysis. Adv. Second Messenger Phosphoprot. Res. 1988, 22, 175–191. [Google Scholar]
- Jitrapakdee, S. Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 2012, 44, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Lent, B.A.; Kim, K.H. Phosphorylation and activation of acetyl-coenzyme A carboxylase kinase by the catalytic subunit of cyclic AMP-dependent protein kinase. Arch. Biochem. Biophys. 1983, 225, 972–978. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Williams, K.W.; Gautron, L.; Elmquist, J.K. Induction of leptin resistance by activation of cAMP-Epac signaling. Cell Metab. 2011, 13, 331–339. [Google Scholar] [CrossRef]
- Yan, J.; Mei, F.C.; Cheng, H.; Lao, D.H.; Hu, Y.; Wei, J.; Patrikeev, I.; Hao, D.; Stutz, S.J.; Dineley, K.T.; et al. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol. Cell Biol. 2013, 33, 918–926. [Google Scholar] [CrossRef]
- Almahariq, M.; Mei, F.C.; Cheng, X. Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends Endocrinol. Metab. 2014, 25, 60–71. [Google Scholar] [CrossRef]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharm. 2012, 166, 447–456. [Google Scholar] [CrossRef]
- Schippers, M.; Beljaars, L.; Post, E.; Lotersztajn, S.; Reker-Smit, C.; Han, B.; Munoz-Llancao, P.; Schmidt, M.; Poelstra, K. Upregulation of Epac-1 in hepatic stellate cells by prostaglandin E2 in liver fibrosis is associated with reduced fibrogenesis. J. Pharm. Exp. 2017, 363, 126–135. [Google Scholar] [CrossRef]
- Rehmann, H.; Arias-Palomo, E.; Hadders, M.A.; Schwede, F.; Llorca, O.; Bos, J.L. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 2008, 455, 124–127. [Google Scholar] [CrossRef]
- Tsalkova, T.; Blumenthal, D.K.; Mei, F.C.; White, M.A.; Cheng, X. Mechanism of Epac activation: Structural and functional analyses of Epac2 hinge mutants with constitutive and reduced activities. J. Biol. Chem. 2009, 284, 23644–23651. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tsalkova, T.; White, M.A.; Mei, F.C.; Liu, T.; Wang, D.; Woods, V.L., Jr.; Cheng, X. Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J. Biol. Chem. 2011, 286, 17889–17897. [Google Scholar] [CrossRef] [PubMed]
- Consonni, S.V.; Gloerich, M.; Spanjaard, E.; Bos, J.L. cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 3814–3819. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, E.E.; Kolesnikov, S.S.; Lyubarsky, A.L. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature 1985, 313, 310–313. [Google Scholar] [CrossRef]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [Green Version]
- Schindler, R.F.; Brand, T. The Popeye domain containing protein family—A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Subbaraya, I.; Yamamoto, N.S.; Baehr, W.; Kraus-Friedmann, N. Expression of photoreceptor cyclic nucleotide-gated cation channel alpha subunit (CNGC) in the liver and skeletal muscle. FEBS Lett. 1996, 395, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Cook, N.J.; Molday, L.; Reid, D.; Kaupp, U.B.; Molday, R.S. The cGMP-gated channel of bovine rod photoreceptors is localized exclusively in the plasma membrane. J. Biol. Chem. 1989, 264, 6996–6999. [Google Scholar]
- Gevers, W.; Krebs, H.A. The effects of adenine nucleotides on carbohydrate metabolism in pigeon-liver homogenates. Biochem. J. 1996, 98, 720–735. [Google Scholar]
- Khoo, J.C.; Steinberg, D. Stimulation of rat liver phosphorylase kinase by micromolar concentration of Ca2+. FEBS Lett. 1975, 57, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, E.; Ebashi, S. Requirement of Ca ion for the stimulating effect of cyclic 3’,5’-AMP on muscle phosphorylase b kinase. J. Biochem. 1967, 62, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Robb-Gaspers, L.D.; Burnett, P.; Rutter, G.A.; Denton, R.M.; Rizzuto, R.; Thomas, A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann, N. Activation of protein kinase(s) by glucagon and cyclic-AMP in the rat liver: Relationship to metabolic effects. Biochim. Biophys. Acta 1976, 428, 495–508. [Google Scholar] [CrossRef]
- Matsui, S.; Murakami, E.; Takekoshi, N.; Emoto, J.; Matoba, M. Efficacy of dibutyryl cyclic AMP in heart failure unresponsive to catecholamines. Clin. Ther. 1987, 9, 488–499. [Google Scholar]
- Asai, J.; Takenaka, H.; Katoh, N.; Kishimoto, S. Dibutyryl cAMP influences endothelial progenitor cell recruitment during wound neovascularization. J. Invest. Derm. 2006, 126, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Rundfeldt, C.; Steckel, H.; Sörensen, T.; Wlaź, P. The stable cyclic adenosine monophosphate analogue, dibutyryl cyclo-adenosine monophosphate (bucladesine), is active in a model of acute skin inflammation. Arch. Derm. Res. 2012, 304, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Aroonsakool, N.; Yokoyama, U.; Patel, H.H.; Insel, P.A. Increase in cellular cyclic AMP concentrations reverses the profibrogenic phenotype of cardiac myofibroblasts: A novel therapeutic approach for cardiac fibrosis. Mol. Pharm. 2013, 84, 787–793. [Google Scholar] [CrossRef] [Green Version]
- El-Agroudy, N.N.; El-Naga, R.N.; El-Razeq, R.A.; El-Demerdash, E. Forskolin, a hedgehog signalling inhibitor, attenuates carbon tetrachloride-induced liver fibrosis in rats. Br. J. Pharm. 2016, 173, 3248–3260. [Google Scholar] [CrossRef] [Green Version]
- Vetrugno, M.; Uva, M.G.; Russo, V.; Iester, M.; Ciancaglini, M.; Brusini, P.; Centofanti, M.; Rossetti, L.M. Oral administration of forskolin and rutin contributes to intraocular pressure control in primary open angle glaucoma patients under maximum tolerated medical therapy. J. Ocul Pharm. 2012, 28, 536–541. [Google Scholar] [CrossRef]
- Rios-Hoyo, A.; Gutierrez-Salmean, G. New dietary supplements for obesity: What we currently know. Curr. Obes. Rep. 2016, 5, 262–270. [Google Scholar] [CrossRef]
- Sapio, L.; Gallo, M.; Illiano, M.; Chiosi, E.; Naviglio, D.; Spina, A.; Naviglio, S. The natural cAMP elevating compound Forskolin in Cancer therapy: Is it time? J. Cell Physiol 2017, 232, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, H.; Oue, T.; Uehara, S.; Fukuzawa, M. Forskolin, a Hedgehog signal inhibitor, inhibits cell proliferation and induces apoptosis in pediatric tumor cell lines. Mol. Med. Rep. 2010, 3, 133–139. [Google Scholar] [PubMed] [Green Version]
- Makinodan, E.; Marneros, A.G. Protein kinase A activation inhibits oncogenic Sonic hedgehog signalling and suppresses basal cell carcinoma of the skin. Exp. Derm. 2012, 21, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.J.; Woo, N.R.; Shin, I.C.; Kim, S.G. H89, an inhibitor of PKA and MSK, inhibits cyclic-AMP response element binding protein-mediated MAPK phosphatase-1 induction by lipopolysaccharide. Inflamm. Res. 2009, 58, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Muñoz, M.D.; Osma-García, I.C.; Fresno, M.; Iñiguez, M.A. Involvement of PGE2 and the cAMP signalling pathway in the up-regulation of COX-2 and mPGES-1 expression in LPS-activated macrophages. Biochem. J. 2012, 443, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Liu, T.; Chen, Y.; Li, Y.; Li, W. Combination therapy with protein kinase inhibitor H89 and Tetrandrine elicits enhanced synergistic antitumor efficacy. J. Exp. Clin. Cancer Res. 2018, 37, 114. [Google Scholar] [CrossRef]
- Ma, J.; Chen, M.; Xia, S.K.; Shu, W.; Guo, Y.; Wang, Y.H.; Xu, Y.; Bai, X.M.; Zhang, L.; Zhang, H.; et al. Prostaglandin E2 promotes liver cancer cell growth by the upregulation of FUSE-binding protein 1 expression. Int. J. Oncol. 2013, 42, 1093–1104. [Google Scholar] [CrossRef]
- Enserink, J.M.; Christensen, A.E.; de Rooij, J.; van Triest, M.; Schwede, F.; Genieser, H.G.; Døskeland, S.O.; Blank, J.L.; Bos, J.L. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 2002, 4, 901–906. [Google Scholar] [CrossRef]
- Shariati, B.; Thompson, E.L.; Nicol, G.D.; Vasko, M.R. Epac activation sensitizes rat sensory neurons through activation of Ras. Mol. Cell Neurosci. 2016, 70, 54–67. [Google Scholar] [CrossRef]
- Tsalkova, T.; Mei, F.C.; Li, S.; Chepurny, O.G.; Leech, C.A.; Liu, T.; Holz, G.G.; Woods, V.L., Jr.; Cheng, X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc. Natl. Acad. Sci. USA 2012, 109, 18613–18618. [Google Scholar] [CrossRef] [Green Version]
- Almahariq, M.; Chao, C.; Mei, F.C.; Hellmich, M.R.; Patrikeev, I.; Motamedi, M.; Cheng, X. Pharmacological inhibition and genetic knockdown of exchange protein directly activated by cAMP 1 reduce pancreatic cancer metastasis in vivo. Mol. Pharm. 2015, 87, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishige, K.; Kotera, J.; Michibata, H.; Yuasa, K.; Takebayashi, S.; Okumura, K.; Omori, K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J. Biol. Chem. 1999, 274, 18438–18445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross-Langenhoff, M.; Hofbauer, K.; Weber, J.; Schultz, A.; Schultz, J.E. cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J. Biol. Chem. 2006, 281, 2841–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr. Pharm. Des. 2010, 16, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D.; Baillie, G.S.; Maurice, D.H. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: A molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res. 2007, 100, 950–966. [Google Scholar] [CrossRef]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as target of the intracellular signlling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharm. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [Green Version]
- Marivet, M.C.; Bourguignon, J.J.; Lugnier, C.; Mann, A.; Stoclet, J.C.; Wermuth, C.G. Inhibition of cyclic adenosine-3’,5’-monophosphate phosphodiesterase from vascular smooth muscle by rolipram analogues. J. Med. Chem 1989, 32, 1450–1457. [Google Scholar] [CrossRef]
- Drees, M.; Zimmermann, R.; Eisembrand, G. 3′,5′-cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res. 1993, 53, 3058–3061. [Google Scholar]
- Spina, D. PDE4 inhibitors: Current status. Br. J. Pharm. 2008, 155, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Brullo, C.; Massa, M.; Rocca, M.; Rotolo, C.; Guariento, S.; Rivera, D.; Ricciarelli, R.; Fedele, E.; Fossa, P.; Bruno, O. Synthesis, biological evaluation, and molecular modeling of new 3-(cyclopentyloxy)-4-methoxybenzaldehyde O-(2-(2,6-dimethylmorpholino)-2-oxoethyl) Oxime (GEBR-7b) related phosphodiesterase 4D (PDE4D) inhibitors. J. Med. Chem. 2014, 57, 7061–7072. [Google Scholar] [CrossRef]
- Bruno, O.; Romussi, A.; Spallarossa, A.; Brullo, C.; Schenone, S.; Bondavalli, F.; Vanthuyne, N.; Roussel, C. New selective phosphodiesterase 4D inhibitors differently acting on long, short, and supershort isoforms. J. Med. Chem. 2009, 52, 6546–6557. [Google Scholar] [CrossRef] [PubMed]
- Bruno, O.; Fedele, E.; Prickaerts, J.; Parker, L.A.; Canepa, E.; Brullo, C.; Cavallero, A.; Gardella, E.; Balbi, A.; Domenicotti, C.; et al. GEBR-7b, a novel PDE4D selective inhibitor that improves memory in rodents at non-emetic doses. Br. J. Pharm. 2011, 164, 2054–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterniti, I.; Mazzon, E.; Gil, C.; Impellizzeri, D.; Palomo, V.; Redondo, M.; Perez, D.I.; Esposito, E.; Martinez, A.; Cuzzocrea, S. PDE 7 inhibitors: New potential drugs for the therapy of spinal cord injury. PLoS ONE 2011, 6, e15937. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Shimizu, K.; Narita, M.; Manganiello, V.C.; Tagawa, T. Characterization of phosphodiesterase 3 in human malignant melanoma cell line. Anticancer Res. 2002, 22, 3171–3174. [Google Scholar] [PubMed]
- Yamanaka, Y.; Mammoto, T.; Kirita, T.; Mukai, M.; Mashimo, T.; Sugimura, M.; Kishi, Y.; Nakamura, H. Epinephrine inhibits invasion of oral squamous carcinoma cells by modulating intracellular cAMP. Cancer Lett. 2002, 176, 143–148. [Google Scholar] [CrossRef]
- Baljinnyam, E.; Iwatsubo, K.; Kurotani, R.; Wang, X.; Ulucan, C.; Iwatsubo, M.; Lagunoff, D.; Ishikawa, Y. Epac increases melanoma cell migration by a heparan sulfate-related mechanism. Am. J. Physiol. Cell Physiol. 2009, 297, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Baljinnyam, E.; De Lorenzo, M.S.; Xie, L.H.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Nowycky, M.C.; Iwatsubo, K. Exchange protein directly activated by cyclic AMP increases melanoma cell migration by a Ca2+-dependent mechanism. Cancer Res. 2010, 70, 5607–5617. [Google Scholar] [CrossRef]
- Robichaux, W.G.; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef]
- Misra, U.K.; Pizzo, S.V. Epac1-induced cellular proliferation in prostate cancer cells is mediated by B-Raf/ERK and mTOR signaling cascades. J. Cell. Biochem. 2009, 108, 998–1011. [Google Scholar] [CrossRef] [Green Version]
- Grandoch, M.; Rose, A.; ter Braak, M.; Jendrossek, V.; Rübben, H.; Fischer, J.W.; Schmidt, M.; Weber, A.A. Epac inhibits migration and proliferation of human prostate carcinoma cells. Br. J. Cancer 2009, 101, 2038–2042. [Google Scholar] [CrossRef]
- Lim, J.A.; Juhnn, Y.S. Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp. Mol. Med. 2016, 48, e204. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Juhnn, Y.S. cAMP signaling increases histone deacetylase 8 expression via the Epac2-Rap1A-Akt pathway in H1299 lung cancer cells. Exp. Mol. Med. 2017, 49, e297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitali, E.; Cambiaghi, V.; Spada, A.; Tresoldi, A.; Zerbi, A.; Peverelli, E.; Carnaghi, C.; Mantovani, G.; Lania, A.G. cAMP effects in neuroendocrine tumors: The role of Epac and PKA in cell proliferation and adhesion. Exp. Cell Res. 2015, 339, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.Y.; Lee, G.H.; Lee, M.S.; Kim, H.M.; Lee, J.W. Phosphodiesterase inhibitors control A172 human glioblastoma cell death through cAMP-mediated activation of protein kinase A and Epac1/Rap1 pathways. Life Sci. 2012, 90, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Sapio, L.; Di Maiolo, F.; Illiano, M.; Esposito, A.; Chiosi, E.; Spina, A.; Naviglio, S. Targeting protein kinase A in cancer therapy: An update. EXCLI J. 2014, 13, 843–855. [Google Scholar]
- Cho-Chung, Y.S.; Nesterova, M.V. Tumor reversion: Protein kinase A isozyme switching. Ann. N. Y. Acad. Sci. 2005, 1058, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Nadella, K.S.; Kirschner, L.S. Disruption of protein kinase a regulation causes immortalization and dysregulation of D-type cyclins. Cancer Res. 2005, 65, 10307–10315. [Google Scholar] [CrossRef]
- Nesterova, M.; Yokozaki, H.; McDuffie, E.; Cho-Chung, Y.S. Overexpression of RII beta regulatory subunit of protein kinase A in human colon carcinoma cell induces growth arrest and phenotypic changes that are abolished by site-directed mutation of RII beta. Eur. J. Biochem. 1996, 235, 486–494. [Google Scholar] [CrossRef]
- Marko, D.; Pahlke, G.; Merz, K.H.; Eisenbrand, G. Cyclic 3’,5’-nucleotide phosphodiesterases: Potential targets for anticancer therapy. Chem. Res. Toxicol. 2000, 13, 944–948. [Google Scholar] [CrossRef]
- Lin, D.C.; Xu, L.; Ding, L.W.; Sharma, A.; Liu, L.Z.; Yang, H.; Tan, P.; Vadgama, J.; Karlan, B.Y.; Lester, J.; et al. Genomic and functional characterizations of phosphodiesterase subtype 4D in human cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 6109–6114. [Google Scholar] [CrossRef] [Green Version]
- Mouratidis, P.X.; Colston, K.W.; Bartlett, J.B.; Muller, G.W.; Man, H.W.; Stirling, D.; Dalgleish, A.G. Antiproliferative effects of CC-8062 and CC-8075 in pancreatic cancer cells. Pancreas 2009, 38, 78–84. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Brunton, V.G.; Baillie, G.S.; Leslie, N.R.; Houslay, M.D.; Frame, M.C. Chemoresistant KM12C colon cancer cells are addicted to low cyclic AMP levels in phosphodiesterase 4-regualted compartment via effects on phosphoinositide 3-kinase. Cancer Res. 2007, 67, 5248–5257. [Google Scholar] [CrossRef] [PubMed]
- Campagnolo, L.; Giorgi, M.; Augusti-Tocco, G. Phosphodiesterase specific inhibitors control cell growth of a human neuroepithelioma cell line. J. Neurooncol. 1997, 31, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Marko, D.; Romanakis, K.; Zankl, H.; Fürstenberger, G.; Steinbauer, B.; Eisenbrand, G. Induction of apoptosis by an inhibitor of cAMP-specific PDE in malignant murine carcinoma cells overexpressing PDE activity in comparison to their nonmalignant counterparts. Cell Biochem. Biophys. 1998, 28, 75–101. [Google Scholar] [CrossRef]
- Wagner, B.; Jakobs, S.; Habermeyer, M.; Hippe, F.; Cho-Chung, Y.S.; Eisenbrand, G.; Marko, D. 7-Benzylamino-6-chloro-2-piperazino-4-pyrrolidino-pteridine, a potent inhibitor of cAMP-specific phosphodiesterase, enhancing nuclear protein binding to the CRE consensus sequence in human tumour cells. Biochem. Pharm. 2002, 63, 659–668. [Google Scholar] [CrossRef]
- Powers, G.L.; Hammer, K.D.; Domenech, M.; Frantskevich, K.; Malinowski, R.L.; Bushman, W.; Beebe, D.J.; Marker, P.C. Phosphodiesterase 4D inhibitors limit prostate cancer growth potential. Mol. Cancer Res. 2015, 13, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, M.F.; Faucz, F.R.; Bimpaki, E.; Horvath, A.; Levy, I.; de Alexandre, R.B.; Ahmad, F.; Manganiello, V.; Stratakis, C.A. Clinical and molecular genetics of the phosphodiesterases (PDEs). Endocr Rev. 2014, 35, 195–233. [Google Scholar] [CrossRef]
- Reneerkens, O.A.; Rutten, K.; Steinbusch, H.W.; Blokland, A.; Prickaerts, J. Selective phosphodiesterase inhibitors: A promising target for cognition enhancement. Psychopharmacology 2009, 202, 419–443. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, M.; Modica, A.; Pompili, A.; Pacitti, C.; Gasbarri, A. The induction of cyclic nucleotide phosphodiesterase 4 gene (PDE4D) impairs memory in a water maze task. Behav. Brain Res. 2004, 154, 99–106. [Google Scholar] [CrossRef]
- Soares, L.M.; De Vry, J.; Steinbusch, H.W.M.; Milani, H.; Prickaerts, J.; Weffort de Oliveira, R.M. Rolipram improves cognition, reduces anxiety- and despair-like behaviors and impacts hippocampal neuroplasticity after transient global cerebral ischemia. Neuroscience 2016, 326, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Goldhoff, P.; Warrington, N.M.; Limbrick, D.D., Jr.; Hope, A.; Woerner, B.M.; Jackson, E.; Perry, A.; Piwnica-Worms, D.; Rubin, J.B. Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes brain tumor regression. Clin. Cancer Res. 2008, 14, 7717–7725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, R.; Sun, T.; Warrington, N.M.; Rubin, J.B. Treating brain tumors with PDE4 inhibitors. Trends Pharm. Sci. 2011, 32, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, M.; Caniglia, C.; Scarsella, G.; Augusti-Tocco, G. Characterization of 3′,5′ cyclic nucleotide phosphodiesterase activities of mouse neuroblastoma N18TG2 cells. FEBS Lett. 1993, 324, 76–80. [Google Scholar] [CrossRef] [Green Version]
- Ramezani, S.; Vousooghi, N.; Kapourchali, F.R.; Hadjighasem, M.; Hayat, P.; Amini, N.; Joghataei, M.T. Rolipram potentiates bevacizumab-induced cell death in human glioblastoma stem-like cells. Life Sci. 2017, 173, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Brooks, M.D.; Jackson, E.; Warrington, N.M.; Luo, J.; Forys, J.T.; Taylor, S.; Mao, D.D.; Leonard, J.R.; Kim, A.H.; Piwnica-Worms, D.; et al. PDE7B is a novel, prognostically significant mediator of glioblastoma growth whose expression is regulated by endothelial cells. PLoS ONE 2014, 9, e107397. [Google Scholar] [CrossRef]
- Lee, J.; Choi, Y.H.; Nguyen, P.M.; Kim, J.S.; Lee, S.J.; Trepel, J.B. Cyclic AMP induces inhibition of cyclin A expression and growth arrest in human hepatoma cells. Biochi. Biophis. Acta 1999, 1449, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Tarn, W.Y.; Lai, M.C. Translational control of cyclins. Cell Div. 2011, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Paterlini, P.; Flejou, J.F.; De Mitri, M.S.; Pisi, E.; Franco, D.; Bréchot, C. Structure and expression of the cyclin A gene in human primary liver cancer. Correlation with flow cytometric parameters. J. Hepatol. 1995, 23, 47–52. [Google Scholar] [CrossRef]
- Vigneron, S.; Sundermann, L.; Labbé, J.C.; Pintard, L.; Radulescu, O.; Castro, A.; Lorca, T. Cyclin A-cdk1-Dependent phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Dev. Cell 2018, 45, 637–650. [Google Scholar] [CrossRef]
- Chao, Y.; Shih, Y.L.; Chiu, J.H.; Chau, G.Y.; Lui, W.Y.; Yang, W.K.; Lee, S.D.; Huang, T.S. Overexpression of cyclin A but not Skp 2 correlates with the tumor relapse of human hepatocellular carcinoma. Cancer Res. 1998, 58, 985–990. [Google Scholar] [PubMed]
- Giorgi, M.; Leonetti, C.; Citro, G.; Augusti-Tocco, G. In vitro and in vivo inhibition of SK-N-MC neuroblastoma growth using cyclic nucleotide phosphodiesterase inhibitors. J. Neurooncol. 2001, 51, 25–31. [Google Scholar] [CrossRef]
- Fleischhacker, W.W.; Hinterhuber, H.; Bauer, H.; Pflug, B.; Berner, P.; Simhandl, C.; WolfGerlach, W.; Jaklitsch, H.; Sastre-y-Hernández, M.; Schmeding-Wiegel, H.; et al. A multicenter double-blind study of three different doses of the new cAMP-phosphodiesterase inhibitor rolipram in patients with major depressive disorder. Neuropsychobiology 1992, 26, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Pearse, D.D.; Hughes, Z.A. PDE4B as a microglia target to reduce neuroinflammation. Glia 2016, 64, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Ionta, M.; Rosa, M.C.; Almeida, R.B.; Freitas, V.M.; Rezende-Teixeira, P.; Machado-Santelli, G.M. Retinoic acid and cAMP inhibit rat hepatocellular carcinoma cell proliferation and enhance cell differentiation. Braz J. Med. Biol. Res. 2012, 45, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, N. Between death and survival: Retinoic acid in regulation of apoptosis. Ann. Rev. Nutr. 2010, 30, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Ara, C.; Massimi, M.; Conti Devirgiliis, L. Retinoic acid modulates gap junctional intercellular communication in hepatocytes and hepatoma cells. Cell. Mol. Life Sci. 2002, 59, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Ara, C.; Conti DeVirgiliis, L.; Massimi, M. Influence of retinoic acid on adhesion complexes in human hepatoma cells: A clue to its antiproliferative effects. Cell Commun. Adhes. 2004, 11, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.Y.; Park, S.H.; Yoo, Y.D.; Kim, J.S.; Kim, Y.H. CDK2/4 regulate retinoic acid-induced G1 arrest in hepatocellular carcinoma cells. Hepatol. Res. 2005, 31, 143–152. [Google Scholar] [CrossRef]
- Massimi, M.; Conti Devirgiliis, L. Adhesion to the extracellular matrix is positively regulated by retinoic acid in HepG2 cells. Liver Int. 2007, 27, 128–136. [Google Scholar] [CrossRef]
- Yang, J.; Shi, Q.D.; Song, T.B.; Feng, G.F.; Zang, W.J.; Zong, C.H.; Chang, L. Vasoactive intestinal peptide increases VEGF expression to promote proliferation of brain vascular endothelial cells via the cAMP/PKA pathway after ischemic insult in vitro. Peptides 2013, 42, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Takeba, Y.; Iiri, T.; Ohta, Y.; Ootaki, M.; Watanabe, M.; Watanabe, D.; Koizumi, S.; Otsubo, T.; Matsumoto, N. Vasoactive intestinal peptide increases apoptosis of hepatocellular carcinoma by inhibiting the cAMP/Bcl-xL pathway. Cancer Sci. 2019, 110, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacas, E.; Fernández-Martínez, A.B.; Bajo, A.M.; Sánchez-Chapado, M.; Schally, A.V.; Prieto, J.C.; Carmena, M.J. Vasoactive intestinal peptide (VIP) inhibits human renal cell carcinoma proliferation. Biochim. Biophys. Acta 2012, 1823, 1676–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Quan, H.; Xie, C.; Wang, L.; Hu, Y.; Lou, L. Phosphodiesterase 3/4 inhibitor zardaverine exhibits potent and selective antitumor activity against hepatocellular carcinoma both in vitro and in vivo independently of phosphodiesterase inhibition. PLoS ONE 2014, 9, e90627. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.J.; Houslay, M.D. Action of rolipram on specific PDE4 cAMP phosphodiesterase isoforms and on the phosphorylation of cAMP-response-element-binding protein (CREB) and p38 mitogen-activated protein (MAP) kinase in U937 monocytic cells. Biochem J. 2000, 347, 571–578. [Google Scholar] [CrossRef]
- Inui, M.; Martello, G.; Piccolo, S. MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 2010, 11, 252–263. [Google Scholar] [CrossRef]
- Hwang, H.W.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef]
- Khella, H.W.; White, N.M.; Faragalla, H.; Gabril, M.; Boazak, M.; Dorian, D.; Khalil, B.; Antonios, H.; Bao, T.T.; Pasic, M.D.; et al. Exploring the role of miRNAs in renal cell carcinoma progression and metastasis through bioinformatic and experimental analyses. Tumour Biol. 2012, 33, 131–140. [Google Scholar] [CrossRef]
- Huo, W.; Du, M.; Pan, X.; Zhu, X.; Gao, Y.; Li, Z. miR-203a-3p.1 targets IL-24 to modulate hepatocellular carcinoma cell growth and metastasis. FEBS Open Bio. 2017, 7, 1085–1091. [Google Scholar] [CrossRef]
- Menezes, M.E.; Bhatia, S.; Bhoopathi, P.; Das, S.K.; Emdad, L.; Dasgupta, S.; Dent, P.; Wang, X.Y.; Sarkar, D.; Fisher, P.B. MDA-7/IL-24: Multifunctional cancer killing cytokine. Adv. Exp. Med. Biol. 2014, 818, 127–153. [Google Scholar]
- Chen, L.; Gao, H.; Liang, J.; Qiao, J.; Duan, J.; Shi, H.; Zhen, T.; Li, H.; Zhang, F.; Zhu, Z.; et al. miR-203a-3p promotes colorectal cancer proliferation and migration by targeting PDE4D. Am. J. Cancer Res. 2018, 8, 2387–2401. [Google Scholar] [PubMed]
- Kim, D.U.; Nam, J.; Cha, M.D.; Kim, S.W. Inhibition of phosphodiesterase 4D decreases the malignant properties of DLD 1 colorectal cancer cells by repressing the AKT/mTOR/Myc signaling pathway. Oncol Lett. 2019, 17, 3589–3598. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massimi, M.; Ragusa, F.; Cardarelli, S.; Giorgi, M. Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma. Cells 2019, 8, 1511. https://doi.org/10.3390/cells8121511
Massimi M, Ragusa F, Cardarelli S, Giorgi M. Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma. Cells. 2019; 8(12):1511. https://doi.org/10.3390/cells8121511
Chicago/Turabian StyleMassimi, Mara, Federica Ragusa, Silvia Cardarelli, and Mauro Giorgi. 2019. "Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma" Cells 8, no. 12: 1511. https://doi.org/10.3390/cells8121511
APA StyleMassimi, M., Ragusa, F., Cardarelli, S., & Giorgi, M. (2019). Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma. Cells, 8(12), 1511. https://doi.org/10.3390/cells8121511